Sharpless asymmetric dihydroxylation is the chemical reaction of an alkene with osmium tetroxide in the presence of a chiral quinine ligand to form a vicinal diol. The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand. Asymmetric dihydroxylation reactions are also highly site selective, providing products derived from reaction of the most electron-rich double bond in the substrate.

In organic chemistry, an epoxide is a cyclic ether, where the ether forms a three-atom ring: two atoms of carbon and one atom of oxygen. This triangular structure has substantial ring strain, making epoxides highly reactive, more so than other ethers. They are produced on a large scale for many applications. In general, low molecular weight epoxides are colourless and nonpolar, and often volatile.
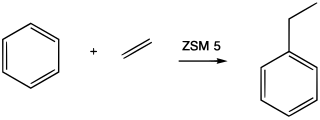
Alkylation is a chemical reaction that entails transfer of an alkyl group. The alkyl group may be transferred as an alkyl carbocation, a free radical, a carbanion, or a carbene. Alkylating agents are reagents for effecting alkylation. Alkyl groups can also be removed in a process known as dealkylation. Alkylating agents are often classified according to their nucleophilic or electrophilic character. In oil refining contexts, alkylation refers to a particular alkylation of isobutane with olefins. For upgrading of petroleum, alkylation produces a premium blending stock for gasoline. In medicine, alkylation of DNA is used in chemotherapy to damage the DNA of cancer cells. Alkylation is accomplished with the class of drugs called alkylating antineoplastic agents.

In organic chemistry, an imine is a functional group or organic compound containing a carbon–nitrogen double bond. The nitrogen atom can be attached to a hydrogen or an organic group (R). The carbon atom has two additional single bonds. Imines are common in synthetic and naturally occurring compounds and they participate in many reactions.
In chemistry, dehydrogenation is a chemical reaction that involves the removal of hydrogen, usually from an organic molecule. It is the reverse of hydrogenation. Dehydrogenation is important, both as a useful reaction and a serious problem. At its simplest, it's a useful way of converting alkanes, which are relatively inert and thus low-valued, to olefins, which are reactive and thus more valuable. Alkenes are precursors to aldehydes, alcohols, polymers, and aromatics. As a problematic reaction, the fouling and inactivation of many catalysts arises via coking, which is the dehydrogenative polymerization of organic substrates.

In organic chemistry, olefin metathesis is an organic reaction that entails the redistribution of fragments of alkenes (olefins) by the scission and regeneration of carbon-carbon double bonds. Because of the relative simplicity of olefin metathesis, it often creates fewer undesired by-products and hazardous wastes than alternative organic reactions. For their elucidation of the reaction mechanism and their discovery of a variety of highly active catalysts, Yves Chauvin, Robert H. Grubbs, and Richard R. Schrock were collectively awarded the 2005 Nobel Prize in Chemistry.
In organic chemistry, hydroboration refers to the addition of a hydrogen-boron bond to certain double and triple bonds involving carbon. This chemical reaction is useful in the organic synthesis of organic compounds.

Tantalum(V) chloride, also known as tantalum pentachloride, is an inorganic compound with the formula TaCl5. It takes the form of a white powder and is commonly used as a starting material in tantalum chemistry. It readily hydrolyzes to form tantalum(V) oxychloride (TaOCl3) and eventually tantalum pentoxide (Ta2O5); this requires that it be synthesised and manipulated under anhydrous conditions, using air-free techniques.

Organoaluminium chemistry is the study of compounds containing bonds between carbon and aluminium. It is one of the major themes within organometallic chemistry. Illustrative organoaluminium compounds are the dimer trimethylaluminium, the monomer triisobutylaluminium, and the titanium-aluminium compound called Tebbe's reagent. The behavior of organoaluminium compounds can be understood in terms of the polarity of the C−Al bond and the high Lewis acidity of the three-coordinated species. Industrially, these compounds are mainly used for the production of polyolefins.
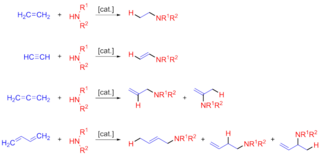
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.
Selenoxide elimination is a method for the chemical synthesis of alkenes from selenoxides. It is most commonly used to synthesize α,β-unsaturated carbonyl compounds from the corresponding saturated analogues. It is mechanistically related to the Cope reaction.
Organoiron chemistry is the chemistry of iron compounds containing a carbon-to-iron chemical bond. Organoiron compounds are relevant in organic synthesis as reagents such as iron pentacarbonyl, diiron nonacarbonyl and disodium tetracarbonylferrate. While iron adopts oxidation states from Fe(−II) through to Fe(VII), Fe(IV) is the highest established oxidation state for organoiron species. Although iron is generally less active in many catalytic applications, it is less expensive and "greener" than other metals. Organoiron compounds feature a wide range of ligands that support the Fe-C bond; as with other organometals, these supporting ligands prominently include phosphines, carbon monoxide, and cyclopentadienyl, but hard ligands such as amines are employed as well.
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing, methods for cleaving the sulfur–carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.

Hydrogen auto-transfer, also known as borrowing hydrogen, is the activation of a chemical reaction by temporary transfer of two hydrogen atoms from the reactant to a catalyst and return of those hydrogen atoms back to a reaction intermediate to form the final product. Two major classes of borrowing hydrogen reactions exist: (a) those that result in hydroxyl substitution, and (b) those that result in carbonyl addition. In the former case, alcohol dehydrogenation generates a transient carbonyl compound that is subject to condensation followed by the return of hydrogen. In the latter case, alcohol dehydrogenation is followed by reductive generation of a nucleophile, which triggers carbonyl addition. As borrowing hydrogen processes avoid manipulations otherwise required for discrete alcohol oxidation and the use of stoichiometric organometallic reagents, they typically display high levels of atom-economy and, hence, are viewed as examples of Green chemistry.
In chemistry, metal-catalysed hydroboration is a reaction used in organic synthesis. It is one of several examples of homogeneous catalysis.
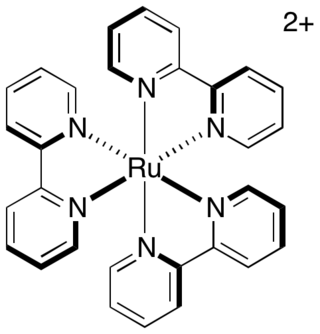
Photoredox catalysis is a branch of photochemistry that uses single-electron transfer. Photoredox catalysts are generally drawn from three classes of materials: transition-metal complexes, organic dyes, and semiconductors. While organic photoredox catalysts were dominant throughout the 1990s and early 2000s, soluble transition-metal complexes are more commonly used today.

Organotantalum chemistry is the chemistry of chemical compounds containing a carbon-to-tantalum chemical bond. A wide variety of compound have been reported, initially with cyclopentadienyl and CO ligands. Oxidation states vary from Ta(V) to Ta(-I).
In organic chemistry, the Murai reaction is an organic reaction that uses C-H activation to create a new C-C bond between a terminal or strained internal alkene and an aromatic compound using a ruthenium catalyst. The reaction, named after Shinji Murai, was first reported in 1993. While not the first example of C-H activation, the Murai reaction is notable for its high efficiency and scope. Previous examples of such hydroarylations required more forcing conditions and narrow scope.
Organoniobium chemistry is the chemistry of compounds containing niobium-carbon (Nb-C) bonds. Compared to the other group 5 transition metal organometallics, the chemistry of organoniobium compounds most closely resembles that of organotantalum compounds. Organoniobium compounds of oxidation states +5, +4, +3, +2, +1, 0, -1, and -3 have been prepared, with the +5 oxidation state being the most common.

Jiro Tsuji was a Japanese chemist, notable for his discovery of organometallic reactions, including the Tsuji-Trost reaction, the Tsuji-Wilkinson decarbonylation, and the Tsuji-Wacker reaction.




