Alpha-synuclein(aSyn) is a protein that, in humans, is encoded by the SNCA gene. Alpha-synuclein is a neuronal protein that regulates synaptic vesicle trafficking and subsequent neurotransmitter release.
Degenerative disease is the result of a continuous process based on degenerative cell changes, affecting tissues or organs, which will increasingly deteriorate over time.

In excitotoxicity, nerve cells suffer damage or death when the levels of otherwise necessary and safe neurotransmitters such as glutamate become pathologically high, resulting in excessive stimulation of receptors. For example, when glutamate receptors such as the NMDA receptor or AMPA receptor encounter excessive levels of the excitatory neurotransmitter, glutamate, significant neuronal damage might ensue. Excess glutamate allows high levels of calcium ions (Ca2+) to enter the cell. Ca2+ influx into cells activates a number of enzymes, including phospholipases, endonucleases, and proteases such as calpain. These enzymes go on to damage cell structures such as components of the cytoskeleton, membrane, and DNA. In evolved, complex adaptive systems such as biological life it must be understood that mechanisms are rarely, if ever, simplistically direct. For example, NMDA in subtoxic amounts induces neuronal survival of otherwise toxic levels of glutamate.
Molecular neuroscience is a branch of neuroscience that observes concepts in molecular biology applied to the nervous systems of animals. The scope of this subject covers topics such as molecular neuroanatomy, mechanisms of molecular signaling in the nervous system, the effects of genetics and epigenetics on neuronal development, and the molecular basis for neuroplasticity and neurodegenerative diseases. As with molecular biology, molecular neuroscience is a relatively new field that is considerably dynamic.

Neuroprotection refers to the relative preservation of neuronal structure and/or function. In the case of an ongoing insult the relative preservation of neuronal integrity implies a reduction in the rate of neuronal loss over time, which can be expressed as a differential equation. It is a widely explored treatment option for many central nervous system (CNS) disorders including neurodegenerative diseases, stroke, traumatic brain injury, spinal cord injury, and acute management of neurotoxin consumption. Neuroprotection aims to prevent or slow disease progression and secondary injuries by halting or at least slowing the loss of neurons. Despite differences in symptoms or injuries associated with CNS disorders, many of the mechanisms behind neurodegeneration are the same. Common mechanisms of neuronal injury include decreased delivery of oxygen and glucose to the brain, energy failure, increased levels in oxidative stress, mitochondrial dysfunction, excitotoxicity, inflammatory changes, iron accumulation, and protein aggregation. Of these mechanisms, neuroprotective treatments often target oxidative stress and excitotoxicity—both of which are highly associated with CNS disorders. Not only can oxidative stress and excitotoxicity trigger neuron cell death but when combined they have synergistic effects that cause even more degradation than on their own. Thus limiting excitotoxicity and oxidative stress is a very important aspect of neuroprotection. Common neuroprotective treatments are glutamate antagonists and antioxidants, which aim to limit excitotoxicity and oxidative stress respectively.
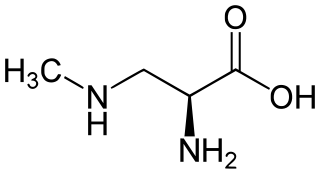
β-Methylamino-L-alanine, or BMAA, is a non-proteinogenic amino acid produced by cyanobacteria. BMAA is a neurotoxin. Its potential role in various neurodegenerative disorders is the subject of scientific research.

In medicine, proteinopathy, or proteopathy, protein conformational disorder, or protein misfolding disease, is a class of diseases in which certain proteins become structurally abnormal, and thereby disrupt the function of cells, tissues and organs of the body. Often the proteins fail to fold into their normal configuration; in this misfolded state, the proteins can become toxic in some way or they can lose their normal function. The proteinopathies include such diseases as Creutzfeldt–Jakob disease and other prion diseases, Alzheimer's disease, Parkinson's disease, amyloidosis, multiple system atrophy, and a wide range of other disorders. The term proteopathy was first proposed in 2000 by Lary Walker and Harry LeVine.

Vacuolar protein sorting ortholog 35 (VPS35) is a protein involved in autophagy and is implicated in neurodegenerative diseases, such as Parkinson's disease (PD) and Alzheimer's disease (AD). VPS35 is part of a complex called the retromer, which is responsible for transporting select cargo proteins between vesicular structures and the Golgi apparatus. Mutations in the VPS35 gene (VPS35) cause aberrant autophagy, where cargo proteins fail to be transported and dysfunctional or unnecessary proteins fail to be degraded. There are numerous pathways affected by altered VPS35 levels and activity, which have clinical significance in neurodegeneration. There is therapeutic relevance for VPS35, as interventions aimed at correcting VPS35 function are in speculation.

Brain mitochondrial carrier protein 1 is a protein that in humans is encoded by the SLC25A14 gene.

Transmembrane protein 106B is a protein that is encoded by the TMEM106B gene. It is found primarily within neurons and oligodendrocytes in the central nervous system with its subcellular location being in lysosomal membranes. TMEM106B helps facilitate important functions for maintaining a healthy lysosome, and therefore certain mutations and polymorphisms can lead to issues with proper lysosomal function. Lysosomes are in charge of clearing out mis-folded proteins and other debris, and thus, play an important role in neurodegenerative diseases that are driven by the accumulation of various mis-folded proteins and aggregates. Due to its impact on lysosomal function, TMEM106B has been investigated and found to be associated to multiple neurodegenerative diseases.
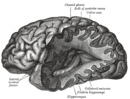
Nervous system diseases, also known as nervous system or neurological disorders, refers to a small class of medical conditions affecting the nervous system. This category encompasses over 600 different conditions, including genetic disorders, infections, cancer, seizure disorders, conditions with a cardiovascular origin, congenital and developmental disorders, and degenerative disorders.
Neuroinflammation is inflammation of the nervous tissue. It may be initiated in response to a variety of cues, including infection, traumatic brain injury, toxic metabolites, or autoimmunity. In the central nervous system (CNS), including the brain and spinal cord, microglia are the resident innate immune cells that are activated in response to these cues. The CNS is typically an immunologically privileged site because peripheral immune cells are generally blocked by the blood–brain barrier (BBB), a specialized structure composed of astrocytes and endothelial cells. However, circulating peripheral immune cells may surpass a compromised BBB and encounter neurons and glial cells expressing major histocompatibility complex molecules, perpetuating the immune response. Although the response is initiated to protect the central nervous system from the infectious agent, the effect may be toxic and widespread inflammation as well as further migration of leukocytes through the blood–brain barrier may occur.

Neurodegenerative diseases can disrupt the normal human homeostasis and result in abnormal estrogen levels. For example, neurodegenerative diseases can cause different physiological effects in males and females. In particular, estrogen studies have revealed complex interactions with neurodegenerative diseases. Estrogen was initially proposed to be a possible treatment for certain types of neurodegenerative diseases but a plethora of harmful side effects such as increased susceptibility to breast cancer and coronary heart disease overshadowed any beneficial outcomes. On the other hand, Estrogen Replacement Therapy has shown some positive effects with postmenopausal women. Estrogen and estrogen-like molecules form a large family of potentially beneficial alternatives that can have dramatic effects on human homeostasis and disease. Subsequently, large-scale efforts were initiated to screen for useful estrogen family molecules. Furthermore, scientists discovered new ways to synthesize estrogen-like compounds that can avoid many side effects.
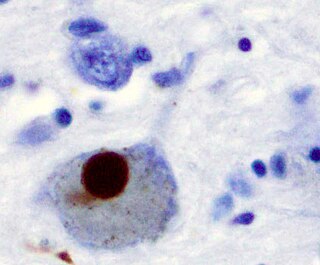
Synucleinopathies are neurodegenerative diseases characterised by the abnormal accumulation of aggregates of alpha-synuclein protein in neurons, nerve fibres or glial cells. There are three main types of synucleinopathy: Parkinson's disease (PD), dementia with Lewy bodies (DLB), and multiple system atrophy (MSA). Other rare disorders, such as various neuroaxonal dystrophies, also have α-synuclein pathologies. Additionally, autopsy studies have shown that around 6% of sporadic Alzheimer's Disease exhibit α-synuclein positive Lewy pathology, and are sub-classed as Alzheimer's Disease with Amygdalar Restricted Lewy Bodies (AD/ALB).

Neurodegenerative diseases are a heterogeneous group of complex disorders linked by the degeneration of neurons in either the peripheral nervous system or the central nervous system. Their underlying causes are extremely variable and complicated by various genetic and/or environmental factors. These diseases cause progressive deterioration of the neuron resulting in decreased signal transduction and in some cases even neuronal death. Peripheral nervous system diseases may be further categorized by the type of nerve cell affected by the disorder. Effective treatment of these diseases is often prevented by lack of understanding of the underlying molecular and genetic pathology. Epigenetic therapy is being investigated as a method of correcting the expression levels of misregulated genes in neurodegenerative diseases.

The pathophysiology of Parkinson's disease is death of dopaminergic neurons as a result of changes in biological activity in the brain with respect to Parkinson's disease (PD). There are several proposed mechanisms for neuronal death in PD; however, not all of them are well understood. Five proposed major mechanisms for neuronal death in Parkinson's Disease include protein aggregation in Lewy bodies, disruption of autophagy, changes in cell metabolism or mitochondrial function, neuroinflammation, and blood–brain barrier (BBB) breakdown resulting in vascular leakiness.

Virginia Man-Yee Lee is a Chinese-born American biochemist and neuroscientist who specializes in the research of Alzheimer's disease. She is the current John H. Ware 3rd Endowed Professor in Alzheimer's Research at the Department of Pathology and Laboratory Medicine, and the director of the Center for Neurodegenerative Disease Research and co-director of the Marian S. Ware Alzheimer Drug Discovery Program at the Perelman School of Medicine, University of Pennsylvania. She received the 2020 Breakthrough Prize in Life Sciences.
The neuroscience of aging is the study of the changes in the nervous system that occur with ageing. Aging is associated with many changes in the central nervous system, such as mild atrophy of the cortex that is considered non-pathological. Aging is also associated with many neurological and neurodegenerative disease such as amyotrophic lateral sclerosis, dementia, mild cognitive impairment, Parkinson's disease, and Creutzfeldt–Jakob disease.
Ted M. Dawson is an American neurologist and neuroscientist. He is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases and Director of the Institute for Cell Engineering at Johns Hopkins University School of Medicine. He has joint appointments in the Department of Neurology, Neuroscience and Department of Pharmacology and Molecular Sciences.

Animal models of Parkinson's disease are essential in the research field and widely used to study Parkinson's disease. Parkinson's disease is a neurodegenerative disorder, characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc). The loss of the dopamine neurons in the brain, results in motor dysfunction, ultimately causing the four cardinal symptoms of PD: tremor, rigidity, postural instability, and bradykinesia. It is the second most prevalent neurodegenerative disease, following Alzheimer's disease. It is estimated that nearly one million people could be living with PD in the United States.