| Alpha-1 antitrypsin deficiency | |
|---|---|
| Other names | α1-antitrypsin deficiency |
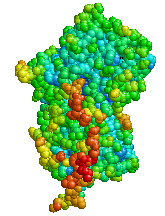 | |
| Structure of Alpha-1 antitrypsin | |
| Specialty | Pulmonology, medical genetics |
| Symptoms | Shortness of breath, wheezing, yellowish skin [1] |
| Complications | COPD, cirrhosis, neonatal jaundice, panniculitis [1] |
| Usual onset | 20 to 50 years old [1] |
| Causes | Mutation in the SERPINA1 gene [1] |
| Diagnostic method | Based on symptoms, blood tests, genetic tests [2] |
| Differential diagnosis | Asthma [1] |
| Treatment | Medications, lung transplant, liver transplant [2] |
| Medication | Bronchodilators, inhaled steroids, antibiotics, intravenous infusions of A1AT protein [2] |
| Prognosis | Life expectancy ~50 years (smokers), nearly normal (non smokers) [3] |
| Frequency | 1 in 2,500 (Europeans) [1] |
Alpha-1 antitrypsin deficiency (A1AD or AATD) is a genetic disorder that may result in lung disease or liver disease. [1] Onset of lung problems is typically between 20 and 50 years old. [1] This may result in shortness of breath, wheezing, or an increased risk of lung infections. [1] [2] Complications may include COPD, cirrhosis, neonatal jaundice, or panniculitis. [1]

A genetic disorder is a genetic problem caused by one or more abnormalities formed in the genome. Most genetic disorders are quite rare and affect one person in every several thousands or millions. The earliest known genetic condition in a hominid was in the fossil species Paranthropus robustus, with over a third of individuals displaying Amelogenesis imperfecta.

Liver disease is a type of damage to or disease of the liver. Whenever the course of the problem lasts long, chronic liver disease ensues.
Shortness of breath, also known as dyspnea, is the feeling that one cannot breathe well enough. The American Thoracic Society defines it as "a subjective experience of breathing discomfort that consists of qualitatively distinct sensations that vary in intensity", and recommends evaluating dyspnea by assessing the intensity of the distinct sensations, the degree of distress involved, and its burden or impact on activities of daily living. Distinct sensations include effort/work, chest tightness, and air hunger.
Contents
- Signs and symptoms
- Associated conditions
- Genetics
- Pathophysiology
- Diagnosis
- Treatment
- A1AT protein
- Epidemiology
- History
- Research
- References
- External links
A1AD is due to a mutation in the SERPINA1 gene that results in not enough alpha-1 antitrypsin (A1AT). [1] Risk factors for lung disease include cigarette smoking and environmental dust. [1] The underlying mechanism involves unblocked neutrophil elastase and build up of abnormal A1AT in the liver. [1] It is autosomal co-dominant, meaning that one defective allele tends to result in milder disease than two defective alleles. [1] The diagnosis is suspected based on symptoms and confirmed by blood tests or genetic tests. [2]
Neutrophil elastase is a serine proteinase in the same family as chymotrypsin and has broad substrate specificity. Secreted by neutrophils and macrophages during inflammation, it destroys bacteria and host tissue. It also localizes to Neutrophil extracellular traps (NETs), via its high affinity for DNA, an unusual property for serine proteases.
An allele is a variant form of a given gene. Sometimes, the presence of different alleles of the same gene can result in different observable phenotypic traits, such as different pigmentation. A notable example of this trait of color variation is Gregor Mendel's discovery that the white and purple flower colors in pea plants were the result of "pure line" traits which could be used as a control for future experiments. However, most genetic variations result in little or no observable variation.
Treatment of lung disease may include bronchodilators, inhaled steroids, and when infections occur antibiotics. [2] Intravenous infusions of the A1AT protein or in severe disease lung transplantation may also be recommended. [2] In those with severe liver disease liver transplantation may be an option. [2] [4] Avoiding smoking is recommended. [2] Vaccination for influenza, pneumococcus, and hepatitis is also recommended. [2] Life expectancy among those who smoke is 50 years old while among those who do not smoke it is almost normal. [3]

Lung transplantation, or pulmonary transplantation, is a surgical procedure in which a patient's diseased lungs are partially or totally replaced by lungs which come from a donor. Donor lungs can be retrieved from a living donor or a deceased donor. A living donor can only donate one lung lobe. With some lung diseases, a recipient may only need to receive a single lung. With other lung diseases such as cystic fibrosis, it is imperative that a recipient receive two lungs. While lung transplants carry certain associated risks, they can also extend life expectancy and enhance the quality of life for end-stage pulmonary patients.

Liver transplantation or hepatic transplantation is the replacement of a diseased liver with the healthy liver from another person (allograft). Liver transplantation is a treatment option for end-stage liver disease and acute liver failure, although availability of donor organs is a major limitation. The most common technique is orthotopic transplantation, in which the native liver is removed and replaced by the donor organ in the same anatomic position as the original liver. The surgical procedure is complex, requiring careful harvest of the donor organ and meticulous implantation into the recipient. Liver transplantation is highly regulated, and only performed at designated transplant medical centers by highly trained transplant physicians and supporting medical team. The duration of the surgery ranges from 4 to 18 hours depending on outcome. Favorable outcomes require careful screening for eligible recipient, as well as a well-calibrated live or cadaveric donor match.

Influenza vaccines, also known as flu shots or flu jabs, are vaccines that protect against infection by influenza viruses. A new version of the vaccine is developed twice a year, as the influenza virus rapidly changes. While their effectiveness varies from year to year, most provide modest to high protection against influenza. The United States Centers for Disease Control and Prevention (CDC) estimates that vaccination against influenza reduces sickness, medical visits, hospitalizations, and deaths. When an immunized worker does catch the flu, they are on average back at work a half day sooner. Vaccine effectiveness in those under two years old and over 65 years old remains unknown due to the low quality of the research. Vaccinating children may protect those around them.
The condition affects about 1 in 2,500 people of European descent. [1] Severe deficiency occurs in about 1 in 5,000. [5] In Asians it is uncommon. [1] About 3% of people with COPD are believed to have the condition. [5] Alpha-1 antitrypsin deficiency was first described in the 1960s. [6]






















