
A brain tumor occurs when abnormal cells form within the brain. There are two main types of tumors: malignant (cancerous) tumors and benign (non-cancerous) tumors. These can be further classified as primary tumors, which start within the brain, and secondary tumors, which most commonly have spread from tumors located outside the brain, known as brain metastasis tumors. All types of brain tumors may produce symptoms that vary depending on the size of the tumor and the part of the brain that is involved. Where symptoms exist, they may include headaches, seizures, problems with vision, vomiting and mental changes. Other symptoms may include difficulty walking, speaking, with sensations, or unconsciousness.
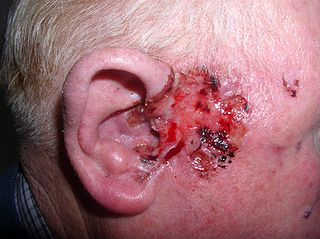
Basal-cell carcinoma (BCC), also known as basal-cell cancer, basalioma or rodent ulcer, is the most common type of skin cancer. It often appears as a painless raised area of skin, which may be shiny with small blood vessels running over it. It may also present as a raised area with ulceration. Basal-cell cancer grows slowly and can damage the tissue around it, but it is unlikely to spread to distant areas or result in death.
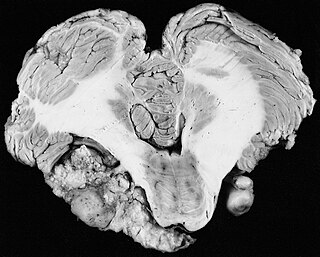
A vestibular schwannoma (VS), also called acoustic neuroma, is a benign tumor that develops on the vestibulocochlear nerve that passes from the inner ear to the brain. The tumor originates when Schwann cells that form the insulating myelin sheath on the nerve malfunction. Normally, Schwann cells function beneficially to protect the nerves which transmit balance and sound information to the brain. However, sometimes a mutation in the tumor suppressor gene, NF2, located on chromosome 22, results in abnormal production of the cell protein named Merlin, and Schwann cells multiply to form a tumor. The tumor originates mostly on the vestibular division of the nerve rather than the cochlear division, but hearing as well as balance will be affected as the tumor enlarges.

Oligodendrogliomas are a type of glioma that are believed to originate from the oligodendrocytes of the brain or from a glial precursor cell. They occur primarily in adults but are also found in children.

An ependymoma is a tumor that arises from the ependyma, a tissue of the central nervous system. Usually, in pediatric cases the location is intracranial, while in adults it is spinal. The common location of intracranial ependymomas is the fourth ventricle. Rarely, ependymomas can occur in the pelvic cavity.

Meningioma, also known as meningeal tumor, is typically a slow-growing tumor that forms from the meninges, the membranous layers surrounding the brain and spinal cord. Symptoms depend on the location and occur as a result of the tumor pressing on nearby tissue. Many cases never produce symptoms. Occasionally seizures, dementia, trouble talking, vision problems, one sided weakness, or loss of bladder control may occur.

Radiosurgery is surgery using radiation, that is, the destruction of precisely selected areas of tissue using ionizing radiation rather than excision with a blade. Like other forms of radiation therapy, it is usually used to treat cancer. Radiosurgery was originally defined by the Swedish neurosurgeon Lars Leksell as "a single high dose fraction of radiation, stereotactically directed to an intracranial region of interest".

A benign tumor is a mass of cells (tumor) that does not invade neighboring tissue or metastasize. Compared to malignant (cancerous) tumors, benign tumors generally have a slower growth rate. Benign tumors have relatively well differentiated cells. They are often surrounded by an outer surface or stay contained within the epithelium. Common examples of benign tumors include moles and uterine fibroids.

Phyllodes tumors, are a rare type of biphasic fibroepithelial mass that form from the periductal stromal and epithelial cells of the breast. They account for less than 1% of all breast neoplasms. They were previously termed cystosarcoma phyllodes, coined by Johannes Müller in 1838, before being renamed to phyllodes tumor by the World Health Organization in 2003. Phullon, which means 'leaf' in Greek, describes the unique papillary projections characteristic of phyllodes tumors on histology. Diagnosis is made via a core-needle biopsy and treatment is typically surgical resection with wide margins (>1 cm), due to their propensity to recur.

A craniopharyngioma is a rare type of brain tumor derived from pituitary gland embryonic tissue that occurs most commonly in children, but also affects adults. It may present at any age, even in the prenatal and neonatal periods, but peak incidence rates are childhood-onset at 5–14 years and adult-onset at 50–74 years. People may present with bitemporal inferior quadrantanopia leading to bitemporal hemianopsia, as the tumor may compress the optic chiasm. It has a point prevalence around two per 1,000,000. Craniopharyngiomas are distinct from Rathke's cleft tumours and intrasellar arachnoid cysts.

Pleomorphic adenoma is a common benign salivary gland neoplasm characterised by neoplastic proliferation of epithelial (ductal) cells along with myoepithelial components, having a malignant potentiality. It is the most common type of salivary gland tumor and the most common tumor of the parotid gland. It derives its name from the architectural Pleomorphism seen by light microscopy. It is also known as "Mixed tumor, salivary gland type", which refers to its dual origin from epithelial and myoepithelial elements as opposed to its pleomorphic appearance.
Schwannomatosis is an extremely rare genetic disorder closely related to the more-common disorder neurofibromatosis (NF). Originally described in Japanese patients, it consists of multiple cutaneous schwannomas, central nervous system tumors, and other neurological complications, excluding hallmark signs of NF. The exact frequency of schwannomatosis cases is unknown, although some populations have noted frequencies as few as 1 case per 1.7 million people.

Hemangioblastomas, or haemangioblastomas, are vascular tumors of the central nervous system that originate from the vascular system, usually during middle age. Sometimes, these tumors occur in other sites such as the spinal cord and retina. They may be associated with other diseases such as polycythemia, pancreatic cysts and Von Hippel–Lindau syndrome. Hemangioblastomas are most commonly composed of stromal cells in small blood vessels and usually occur in the cerebellum, brainstem or spinal cord. They are classed as grade I tumors under the World Health Organization's classification system.

Choroid plexus papilloma, also known as papilloma of the choroid plexus, is a rare benign neuroepithelial intraventricular WHO grade I lesion found in the choroid plexus. It leads to increased cerebrospinal fluid production, thus causing increased intracranial pressure and hydrocephalus.

Dysembryoplastic neuroepithelial tumour is a type of brain tumor. Most commonly found in the temporal lobe, DNTs have been classified as benign tumours. These are glioneuronal tumours comprising both glial and neuron cells and often have ties to focal cortical dysplasia.
Esthesioneuroblastoma is a rare cancer of the nasal cavity. Arising from the upper nasal tract, esthesioneuroblastoma is believed to originate from sensory neuroepithelial cells, also known as neuroectodermal olfactory cells.
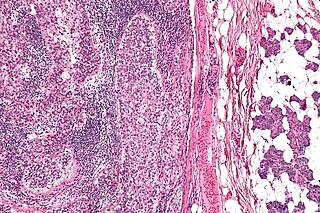
Sebaceous carcinoma, also known as sebaceous gland carcinoma (SGc), sebaceous cell carcinoma, and meibomian gland carcinoma is an uncommon malignant cutaneous tumor. Most are typically about 1.4 cm at presentation. SGc originates from sebaceous glands in the skin and, therefore, may originate anywhere in the body where these glands are found. SGc can be divided into 2 types: periocular and extraocular. The periocular region is rich in sebaceous glands making it a common site of origin. The cause of these lesions in the vast majority of cases is unknown. Occasional cases may be associated with Muir-Torre syndrome. SGc accounts for approximately 0.7% of all skin cancers, and the incidence of SGc is highest in Caucasian, Asian, and Indian populations. Due to the rarity of this tumor and variability in clinical and histological presentation, SGc is often misdiagnosed as an inflammatory condition or a more common neoplasm. SGc is commonly treated with wide local excision or Mohs micrographic surgery, and the relative survival rates at 5 and 10 years are 92.72 and 86.98%, respectively.
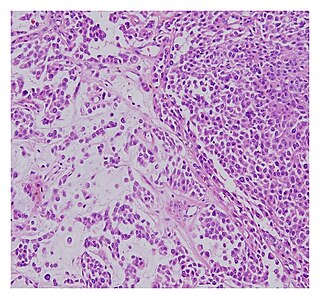
A malignant chondroid syringoma is a very uncommon cutaneous (skin) condition characterised by an adnexal eccrine tumour.
Cardiac fibroma, also known as cardiac fibromatosis, cardiac fibrous hamartoma, fibroelastic hamartoma of heart and fibroma of heart is the second highest type of primary cardiac tumor seen in infants and children. This benign tumor made by connective tissue and fibroblast is largely observed in the ventricles of the heart. The left ventricle is the most common location of cardiac fibroma and accounts for approximately 57% of cardiac fibroma cases followed by the right ventricle with 27.5% of cases. Symptoms of the disease depend on the size of the tumor, its location relative to the conduction system, and whether it obstructs blood flow. Two-thirds of children with this tumor are asymptomatic, showing no signs and symptoms. Therefore the cause of cardiac fibroma is unexplained but has been associated with Gorlin Syndrome. Echocardiography is the primarily diagnostic method used to detect if an individual has cardiac fibroma. Resection of the tumor is recommended however heart transplantation is done if surgery is not possible. Overall prognosis of resection is favorable and the chance of recurrence is scarcely reported.

Astroblastoma is a rare glial tumor derived from the astroblast, a type of cell that closely resembles spongioblastoma and astrocytes. Astroblastoma cells are most likely found in the supratentorial region of the brain that houses the cerebrum, an area responsible for all voluntary movements in the body. It also occurs significantly in the frontal lobe, parietal lobe, and temporal lobe, areas where movement, language creation, memory perception, and environmental surroundings are expressed. These tumors can be present in major brain areas not associated with the main cerebral hemispheres, including the cerebellum, optic nerve, cauda equina, hypothalamus, and brain stem.




