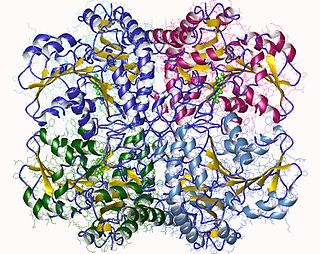
Tryptophan synthase or tryptophan synthetase is an enzyme that catalyses the final two steps in the biosynthesis of tryptophan. It is commonly found in Eubacteria, Archaebacteria, Protista, Fungi, and Plantae. However, it is absent from Animalia. It is typically found as an α2β2 tetramer. The α subunits catalyze the reversible formation of indole and glyceraldehyde-3-phosphate (G3P) from indole-3-glycerol phosphate (IGP). The β subunits catalyze the irreversible condensation of indole and serine to form tryptophan in a pyridoxal phosphate (PLP) dependent reaction. Each α active site is connected to a β active site by a 25 angstrom long hydrophobic channel contained within the enzyme. This facilitates the diffusion of indole formed at α active sites directly to β active sites in a process known as substrate channeling. The active sites of tryptophan synthase are allosterically coupled.
Aminolevulinic acid synthase (ALA synthase, ALAS, or delta-aminolevulinic acid synthase) is an enzyme (EC 2.3.1.37) that catalyzes the synthesis of δ-aminolevulinic acid (ALA) the first common precursor in the biosynthesis of all tetrapyrroles such as hemes, cobalamins and chlorophylls. The reaction is as follows:

Pyridoxal phosphate (PLP, pyridoxal 5'-phosphate, P5P), the active form of vitamin B6, is a coenzyme in a variety of enzymatic reactions. The International Union of Biochemistry and Molecular Biology has catalogued more than 140 PLP-dependent activities, corresponding to ~4% of all classified activities. The versatility of PLP arises from its ability to covalently bind the substrate, and then to act as an electrophilic catalyst, thereby stabilizing different types of carbanionic reaction intermediates.

Aspartate transaminase (AST) or aspartate aminotransferase, also known as AspAT/ASAT/AAT or (serum) glutamic oxaloacetic transaminase, is a pyridoxal phosphate (PLP)-dependent transaminase enzyme that was first described by Arthur Karmen and colleagues in 1954. AST catalyzes the reversible transfer of an α-amino group between aspartate and glutamate and, as such, is an important enzyme in amino acid metabolism. AST is found in the liver, heart, skeletal muscle, kidneys, brain, red blood cells and gall bladder. Serum AST level, serum ALT level, and their ratio are commonly measured clinically as biomarkers for liver health. The tests are part of blood panels.

Triose-phosphate isomerase is an enzyme that catalyzes the reversible interconversion of the triose phosphate isomers dihydroxyacetone phosphate and D-glyceraldehyde 3-phosphate.
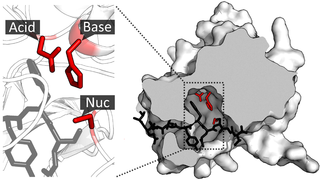
A catalytic triad is a set of three coordinated amino acids that can be found in the active site of some enzymes. Catalytic triads are most commonly found in hydrolase and transferase enzymes. An acid-base-nucleophile triad is a common motif for generating a nucleophilic residue for covalent catalysis. The residues form a charge-relay network to polarise and activate the nucleophile, which attacks the substrate, forming a covalent intermediate which is then hydrolysed to release the product and regenerate free enzyme. The nucleophile is most commonly a serine or cysteine amino acid, but occasionally threonine or even selenocysteine. The 3D structure of the enzyme brings together the triad residues in a precise orientation, even though they may be far apart in the sequence.

Methylmalonyl-CoA mutase (EC 5.4.99.2, MCM), mitochondrial, also known as methylmalonyl-CoA isomerase, is a protein that in humans is encoded by the MUT gene. This vitamin B12-dependent enzyme catalyzes the isomerization of methylmalonyl-CoA to succinyl-CoA in humans. Mutations in MUT gene may lead to various types of methylmalonic aciduria.
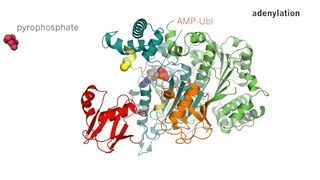
Enzyme catalysis is the increase in the rate of a process by a biological molecule, an "enzyme". Most enzymes are proteins, and most such processes are chemical reactions. Within the enzyme, generally catalysis occurs at a localized site, called the active site.

Serine hydroxymethyltransferase (SHMT) is a pyridoxal phosphate (PLP) (Vitamin B6) dependent enzyme (EC 2.1.2.1) which plays an important role in cellular one-carbon pathways by catalyzing the reversible, simultaneous conversions of L-serine to glycine and tetrahydrofolate (THF) to 5,10-Methylenetetrahydrofolate (5,10-CH2-THF). This reaction provides the largest part of the one-carbon units available to the cell.
Lysine 2,3-aminomutase is a radical SAM enzyme that facilitates the conversion of the amino acid lysine to beta-lysine. It accomplishes this interconversion using three cofactors and a 5'-deoxyadenosyl radical formed in a S-Adenosyl methionine (SAM) activated radical reaction pathway.[1] The generalized reaction is shown below:

Cystathionine-β-synthase, also known as CBS, is an enzyme (EC 4.2.1.22) that in humans is encoded by the CBS gene. It catalyzes the first step of the transsulfuration pathway, from homocysteine to cystathionine:
The enzyme cystathionine γ-lyase (EC 4.4.1.1, CTH or CSE; also cystathionase; systematic name L-cystathionine cysteine-lyase (deaminating; 2-oxobutanoate-forming)) breaks down cystathionine into cysteine, 2-oxobutanoate (α-ketobutyrate), and ammonia:

In enzymology, an alanine racemase is an enzyme that catalyzes the chemical reaction
In enzymology, a beta-lysine 5,6-aminomutase is an enzyme that catalyzes the chemical reaction

Cystathionine beta-lyase, also commonly referred to as CBL or β-cystathionase, is an enzyme that primarily catalyzes the following α,β-elimination reaction

The enzyme methionine γ-lyase (EC 4.4.1.11, MGL) is in the γ-family of PLP-dependent enzymes. It degrades sulfur-containing amino acids to α-keto acids, ammonia, and thiols:

The enzyme Acid-Induced Arginine Decarboxylase (AdiA), also commonly referred to as arginine decarboxylase, catalyzes the conversion of L-arginine into agmatine and carbon dioxide. The process consumes a proton in the decarboxylation and employs a pyridoxal-5'-phosphate (PLP) cofactor, similar to other enzymes involved in amino acid metabolism, such as ornithine decarboxylase and glutamine decarboxylase. It is found in bacteria and virus, though most research has so far focused on forms of the enzyme in bacteria. During the AdiA catalyzed decarboxylation of arginine, the necessary proton is consumed from the cell cytoplasm which helps to prevent the over-accumulation of protons inside the cell and serves to increase the intracellular pH. Arginine decarboxylase is part of an enzymatic system in Escherichia coli, Salmonella Typhimurium, and methane-producing bacteria Methanococcus jannaschii that makes these organisms acid resistant and allows them to survive under highly acidic medium.

The enzyme diaminopimelate decarboxylase (EC 4.1.1.20) catalyzes the cleavage of carbon-carbon bonds in meso 2,6 diaminoheptanedioate to produce CO2 and L-lysine, the essential amino acid. It employs the cofactor pyridoxal phosphate, also known as PLP, which participates in numerous enzymatic transamination, decarboxylation and deamination reactions.

In enzymology, a serine C-palmitoyltransferase (EC 2.3.1.50) is an enzyme that catalyzes the chemical reaction:
Glutamate 2,3-aminomutase is an enzyme that belongs to the radical s-adenosyl methionine (SAM) superfamily. Radical SAM enzymes facilitate the reductive cleavage of S-adenosylmethionine (SAM) through the use of radical chemistry and an iron-sulfur cluster. This enzyme family is implicated in the biosynthesis of DNA precursors, vitamin, cofactor, antibiotic and herbicides and in biodegradation pathways. In particular, glutamate 2,3 aminomutase is involved in the conversion of L-alpha-glutamate to L-beta-glutamate in Clostridium difficile. The generalized reaction is shown below:







