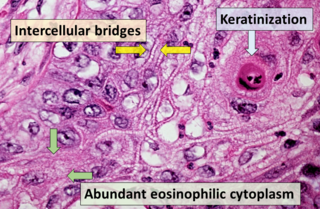
Although rhabdoid variants of LCLC are sometimes referred to as "rhabdoid carcinomas", this particular term should be reserved for examples of "pure" rhabdoid neoplasms (i.e. those that do not contain cells containing other histological variants)
Classification
Lung cancers are now considered a large and extremely heterogeneous family of neoplasms[4] that feature widely varying genetic, biological, and clinical characteristics. About 50 different lung cancer variants are recognized under the 2004 revision of the World Health Organization ("WHO-2004") histological typing system, the most widely recognized and used lung cancer classification scheme.[2] Recent studies have shown beyond doubt that the old classification paradigm of "small cell carcinoma vs. non-small cell carcinoma" is now obsolete, and that the correct "subclassification" of lung cancer cases is necessary to assure that patients receive optimum management.[5][6]
More than 99% of primary lung cancers are carcinoma, which are tumors composed of cells that originate from embryonicectoderm or endoderm, or that feature epithelial characteristics or differentiation.[7] Eight major groups of lung carcinomas are recognized in WHO-2004:[2]
The histogenesis of most lung cancers is not well understood. Carcinomas of the lung are thought to arise from the uncontrolled growth of mutated, transformed, multipotent "cancer stem cells" with epithelial characteristics or lineage. When viewed under a light microscope, the transformed cancer cells in LCLC are undifferentiated, lacking specific cytological and tissue architectural characteristics of other types, subtypes, and variants of lung cancer.[2] Election microscopic studies, however, have shown that many LCLC do have ultrastructural characteristics of other tumor types (i.e. adenocarcinoma, squamous cell carcinoma).,[8] and that rhabdoid carcinomas often show similar features.[9]
Some evidence suggests that cells with the rhabdoid phenotype result from mutations occurring in some of the cells descending from the "parent" tumor, leading to the "emergence" of distinct populations of characteristic cells with the rhabdoid phenotype within the parent neoplasm, often in the peripheral part of the tumor.[10][11][9] Missense mutations occurring in the cytokeratin 8 (CK-8) gene (RTK 8)[12] at specific codons affects the way the protein products of this gene assume their normal two- and three-dimensional shapes, and may well affect the way the mutant proteins undergo assembly into filamentous structures within the cytoplasm. The defective "protofilament" products apparently accumulate aberrantly, and thus form the distinctive whorled paranuclear inclusions that are characteristic of the rhabdoid cell.[13]
It seems likely that mutations and post-translational modifications affecting cytokeratin 8, cytokeratin 18, and vimentin protofilaments are intimately involved in the genesis of the characteristic inclusions and, therefore, of the rhabdoid phenotype. The particulars of this process are poorly understood, but depend in part on the origin of the tumor[14] and stochastic genomic phenomena.[15][16]
Rhabdoid cells often express protein products suggestive of aggressive, dedifferentiated cells, including neuroendocrine tumor-related products[17] and granulocyte-macrophage colony stimulating factor (GM-CSF).[9][18]Vimentin, an intermediate filament protein usually associated with non-carcinomatous tumors (i.e. sarcoma), is ubiquitous in rhabdoid cells.[15] Co-expression of cytokeratins and vimentin are associated with cells undergoing epithelial-mesenchymal transition (EMT).[19][20]
While undifferentiated large-cell lung carcinoma is the most common parent lung tumor from which a rhabdoid phenotype evolves,[21] malignant cells with a rhabdoid phenotype are known to occur in many different histological variants of lung cancer, including adenocarcinoma,[22] sarcomatoid carcinoma,[23][22] squamous cell carcinoma,[24] combined large cell neuroencrine carcinoma,[24] and mucinous bronchioloalveolar carcinoma [25] and combined small cell lung carcinoma.[24]
Metastasis
LCLC-RP is generally considered to be an especially aggressive malignancy that metastasizes widely early on in its clinical course. Similar to most other forms of lung carcinoma, LCLC-RP may spread ("metastasize") in three major ways — by local extension and infiltration into surrounding tissues, by lymphatic spread to regional lymph nodes, and through the bloodstream (hematogeneous metastasis) to distant organs and tissues such as the liver, brain, and skeleton.[citation needed]
It has been reported recently[26] that LCLC-RP can metastasize locally within the airways ("aerogeneous spread"), an uncommon mechanism of extension wherein tumor cells migrate along the lung walls and septa, but do not destroy air sacs. Previously, this type of metastatic behavior had not been seen in this particular tumor, being traditionally associated almost exclusively with the "pneumonic" form of pulmonarybronchioloalveolar carcinoma.[citation needed]
Diagnosis
While occasional scattered rhabdoid cell formation occurs with considerable frequency in lung carcinomas, this is not considered to be of clinical significance.[21][17] According to current classification criteria, a tumor can only be diagnosed as LCLC-RP when an undifferentiated large-cell lung carcinoma contains a rhabdoid cell component that makes up at least 10% of the tumor mass.[27]
Microscopic characteristics of rhabdoid cells include:[2][21][23]
Oval to polygonal cell shape
Eosinophilic, hyaline-like perinuclear agglomerations of intermediate filaments
The differential diagnosis of LCLC-RP includes secondary metastatic lesions, malignant melanoma of the lung with rhabdoid phenotype, mucinous adenocarcinomas (particularly those featuring signet-ring cells), rhabdomyosarcoma, epitheloid angiosarcoma, pleural mesothelioma, and plasmacytoma.[21][27][23][10]
Imaging
On radiological imaging, most cases of LCLC-RP are single "coin lesions" or discrete masses, but cases presenting as multiple nodules throughout the lung have also been noted.[28] Cases with inhomogeneous-appearing consolidation on CT have been reported, as well as aerogenous, lepidic-type spread.[26] Some large, centrally located LCLC-RP have been noted to show signs of gross necrosis and cavitation on imaging studies,[11] as well as being associated with the presence of large bullae.[26]
Immunohistochemistry
Immunohistochemistry is an important factor in diagnosis.[29] Results of immunohistochemistry (IHC) staining in rhabdoid lung cancers tends to reflect the multiphasic nature of these tumors. Typically, markers expressed in LCLC-RP include those seen in "generic" NSCLC's, such as epithelial membrane antigen (EMA, 61%), various cytokeratins (CK's, 80%), and markers associated with the underlying "parent" pulmonary carcinoma. Expression of immunomarkers in the rhabdoid cells, however, have often been noted to be weaker and more diffuse than those in the more differentiated tumor cells.[21][23] They also more frequently express "non-carcinomatous" markers typically associated with "dedifferentiated" neoplasms. Expression of thyroid transcription factor-1 (TTF-1), a commonly used marker for primary lung cancers, appears to be less frequent in rhabdoid carcinomas than in most other histotypes of pulmonary cancers.[22]
Vimentin, an intermediate filament protein usually found in sarcoma, is ubiquitously (nearly 100%) expressed diffusely throughout the cytoplasm of the rhabdoid cells, and is often intermingled with CK's in their whorled inclusions. Some studied have reported that neuroendocrine-related markers (i.e. neuron-specific enolase (NSE), neural cell adhesion molecule (NCAM), chromogranin A (CgA), synaptophysin), are also quite frequently expressed in a significant proportion of rhabdoid cells.[21][17][23]
Treatment
Because LCLC-RP is so rare, no clinical trials have ever been conducted that specifically address treatment of this lung cancer variant. Because LCLC-RP is considered a form of non-small cell lung carcinoma (NSCLC), most physicians adhere to published NSCLC treatment guidelines in rhabdoid carcinoma cases. When possible, radical surgical resection with curative intent is the primary treatment of choice in early stage NSCLC's, and can be administered with or without adjuvant, neoadjuvant, or palliative chemotherapy and/or radiotherapy, depending on the disease stage and performance status of the individual patient.[30]
In numerous clinical trials conducted in NSCLC, several different platinum-based chemotherapy regimens have been shown to be more-or-less equally effective.[30] LCLC's, as a subtype of NSCLC, have traditionally been included in many of these clinical trials, and have been treated like other NSCLC's. More recent trials, however, have shown that some newer agents may have particular effectiveness in prolonging survival of LCLC patients.[5][6]Pemetrexed, in particular, has shown significant reduction in the hazard ratio for death when used in patients with LCLC.[31] Taxane-based (paclitaxel, docetaxel) chemotherapy was shown to induce a complete and sustained response in a liver metastasis in a case of LCC-RP. A later-appearing metastasis within mediastinal lymph nodes in the same case also showed a durable response to a taxane alone.[21]
There have also been reports of rhabdoid carcinomas expressing vascular endothelial growth factor (VEGF),[11] suggesting that targeted molecular therapy with VEGF blocking monoclonal antibodies such as bevacizumab may be active in these variants.[32][33][34] However, evidence suggests that caution must be used when treating a cavitated rhabdoid tumor, one that contains significant components of squamous cell differentiation, or large tumors with containing major blood vessels, due to the potential high risk of life-threatening pulmonary hemorrhage.[32]
A recent study reported a case wherein 2 courses of adjuvant therapy with cisplatin and paclitaxel, followed by oral gefitinib, were used after complete resection. The patient had had no recurrence 34 months later.[18]
As large-volume LCLC-RP may show significant central necrosis and cavitation, prudence dictates that oncologists use extreme caution if contemplating the therapeutic use of bevacizumab, other anti-VEGF compounds, or anti-angiogenesis agents in general, which have been associated with a greatly increased risk of severe hemorrhage and hemoptysis that may be quickly fatal in cavatated pulmonary squamous cell carcinomas. Similar elevated risks have also been noted in tumors located near, or containing, large blood vessels.,[11]
Prognosis
LCLC-RP are considered to be especially aggressive tumors with a dismal prognosis.[5][22][16] Many published cases have shown short survival times after diagnosis.[17][35] Some studies suggest that, as the proportion of rhabdoid cells in the tumor increases, the prognosis tends to worsen,[17] although this is most pronounced when the proportion of rhabdoid cells exceeds 5%.[17] With regard to "parent" neoplasms other than LCLC, adenocarcinomas with rhabdoid features have been reported to have worse prognoses than adenocarcinomas without rhabdoid features,[36] although an "adenocarcinoma with rhabdoid phenotype" tumor variant has not been specifically recognized as a distinct entity under the WHO-2004 classification system.[2]
There are case reports of rhabdoid carcinomas recurring after unusually long periods, which is unusual for a fast-growing, aggressive tumor type. One report described a very early stage patient whose tumor recurred 6 years after initial treatment.[11] Although rapidly progressive, fulminant courses seem to be the rule in this entity, long-term survival has also been noted, even post-metastectomy in late stage, distant metastatic disease.[23]
Epidemiology
Although reliable and comprehensive incidence statistics are nonexistent, LCLC-RP is a rare tumor, with only a few hundred cases described in the scientific literature to date.[21][8] LCLC's made up about 10% of lung cancers in most historical series,[2][7][37][38] equating to approximately 22,000 cases per year in the U.S.[39] Of these LCLC cases, it is estimated that about 1% will eventually develop the rhabdoid phenotype during tumor evolution and progression.[8] In one large series of 902 surgically resected lung cancers, only 3 cases (0.3%) were diagnosed as LCLC-RP. In another highly selected series of large-cell lung carcinoma cases, only 4 of 45 tumors (9%) were diagnosed as the rhabdoid phenotype using the 10% criterion, but another 10 (22%) had at least some rhabdoid cell formation.[17] It appears likely, therefore, that LCLC-RP probably comprises between 0.1% and 1.0% of all lung malignancies.
Similar to nearly all variants of lung carcinoma, large cell lung carcinoma with rhabdoid phenotype appears to be highly related to tobacco smoking. It also appears to be significantly more common in males than in females.[22]
Future research
As the rhabdoid phenotype may exclusively be associated with certain missense mutations in the CK-8 gene (or, possibly, the vimentin gene — see above), it may prove possible to develop specific monoclonal antibodies against certain peptides in these aberrant gene products that may target only rhabdoid cells with these specific mutations. Recent reports suggest that antibodies may be easier to get into the interior of cells than previously thought.[citation needed]
History
Although Colby and colleagues were the first to report a primary lung cancer with a rhabdoid phenotype in a paper published in 1995,[40] cells with these characteristic features had been previously noted in 1978, when they were noted to occur in a rare and extremely aggressive form of kidney cancer that appears almost exclusively in young children called "Wilms tumor".[3]
LCLC-RP were first recognized as a distinct entity under the 3rd (published in 1999) revision of the World Health Organization (WHO) lung tumor histological typing scheme.[41] Its placement in the classification schema went unchanged during the 2004 revision.[2]
Related Research Articles
Adenocarcinoma is a type of cancerous tumor that can occur in several parts of the body. It is defined as neoplasia of epithelial tissue that has glandular origin, glandular characteristics, or both. Adenocarcinomas are part of the larger grouping of carcinomas, but are also sometimes called by more precise terms omitting the word, where these exist. Thus invasive ductal carcinoma, the most common form of breast cancer, is adenocarcinoma but does not use the term in its name—however, esophageal adenocarcinoma does to distinguish it from the other common type of esophageal cancer, esophageal squamous cell carcinoma. Several of the most common forms of cancer are adenocarcinomas, and the various sorts of adenocarcinoma vary greatly in all their aspects, so that few useful generalizations can be made about them.
Carcinoma is a malignancy that develops from epithelial cells. Specifically, a carcinoma is a cancer that begins in a tissue that lines the inner or outer surfaces of the body, and that arises from cells originating in the endodermal, mesodermal or ectodermal germ layer during embryogenesis.
Non-small-cell lung cancer (NSCLC), or non-small-cell lung carcinoma, is any type of epithelial lung cancer other than small-cell lung cancer (SCLC). NSCLC accounts for about 85% of all lung cancers. As a class, NSCLCs are relatively insensitive to chemotherapy, compared to small-cell carcinoma. When possible, they are primarily treated by surgical resection with curative intent, although chemotherapy has been used increasingly both preoperatively and postoperatively.
Adenocarcinoma in situ (AIS) of the lung —previously included in the category of "bronchioloalveolar carcinoma" (BAC)—is a subtype of lung adenocarcinoma. It tends to arise in the distal bronchioles or alveoli and is defined by a non-invasive growth pattern. This small solitary tumor exhibits pure alveolar distribution and lacks any invasion of the surrounding normal lung. If completely removed by surgery, the prognosis is excellent with up to 100% 5-year survival.
Large-cell lung carcinoma (LCLC), or large-cell carcinoma (LCC) in short, is a heterogeneous group of undifferentiated malignant neoplasms that lack the cytologic and architectural features of small cell carcinoma and glandular or squamous differentiation. LCC is categorized as a type of NSCLC which originates from epithelial cells of the lung. LCLC is histologically characterized by the presence of large, undifferentiated cells that lack distinctive features of either squamous cell carcinoma or adenocarcinoma. Typically seen in LCLC tumor cells are abundant pale staining cytoplasm and prominent nucleoli.
Combined small cell lung carcinoma is a form of multiphasic lung cancer that is diagnosed by a pathologist when a malignant tumor, arising from transformed cells originating in lung tissue, contains a component of;small cell lung carcinoma (SCLC), admixed with one components of any histological variant of non-small cell lung carcinoma (NSCLC) in any relative proportion.
Treatment of lung cancer refers to the use of medical therapies, such as surgery, radiation, chemotherapy, immunotherapy, percutaneous ablation, and palliative care, alone or in combination, in an attempt to cure or lessen the adverse impact of malignant neoplasms originating in lung tissue.
Fetal adenocarcinoma (FA) of the lung is a rare subtype of pulmonary adenocarcinoma that exhibits tissue architecture and cell characteristics that resemble fetal lung tissue upon microscopic examination. It is currently considered a variant of solid adenocarcinoma with mucin production.
Epithelial-myoepithelial carcinoma of the lung is a very rare histologic form of malignant epithelial neoplasm ("carcinoma") arising from lung tissue.
Targeted therapy of lung cancer refers to using agents specifically designed to selectively target molecular pathways responsible for, or that substantially drive, the malignant phenotype of lung cancer cells, and as a consequence of this (relative) selectivity, cause fewer toxic effects on normal cells.
HOHMS is the medical acronym for "Higher-Order HistoMolecular Stratification", a term and concept which was first applied to lung cancer research and treatment theory.
Mucinous cystadenocarcinoma of the lung (MCACL) is a very rare malignant mucus-producing neoplasm arising from the uncontrolled growth of transformed epithelial cells originating in lung tissue.
Acinar adenocarcinoma is a histological subtype of gland-forming cancer that is diagnosed when cuboidal and/or columnar shaped malignant cells in the neoplastic tissue form acini and tubules. It is a common form of cancer occurring in the lung and prostate gland.
Sarcomatoid carcinoma of the lung is a term that encompasses five distinct histological subtypes of lung cancer, including (1) pleomorphic carcinoma, (2) spindle cell carcinoma, (3) giant cell carcinoma, (4) carcinosarcoma, or (5) pulmonary blastoma.
Giant-cell carcinoma of the lung (GCCL) is a rare histological form of large-cell lung carcinoma, a subtype of undifferentiated lung cancer, traditionally classified within the non-small-cell lung carcinomas (NSCLC).
Adenosquamous lung carcinoma (AdSqLC) is a biphasic malignant tumor arising from lung tissue that is composed of at least 10% by volume each of squamous cell carcinoma (SqCC) and adenocarcinoma (AdC) cells.
Basaloid squamous cell carcinoma (Bas-SqCC) is an uncommon histological variant of lung cancer composed of cells exhibiting cytological and tissue architectural features of both squamous cell lung carcinoma and basal cell carcinoma.
Large-cell neuroendocrine carcinoma of the lung, or pulmonary large-cell neuroendocrine carcinoma (PLCNC), is a highly malignant neoplasm arising from transformed epithelial cells originating in tissues within the pulmonary tree. It is currently considered to be a subtype of large-cell lung carcinoma.
Basaloid large cell carcinoma of the lung, is a rare histological variant of lung cancer featuring certain distinctive cytological, tissue architectural, and immunohistochemical characteristics and clinical behavior.
Squamous-cell carcinoma (SCC), also known as epidermoid carcinoma, comprises a number of different types of cancer that begin in squamous cells. These cells form on the surface of the skin, on the lining of hollow organs in the body, and on the lining of the respiratory and digestive tracts.
References
↑ Rubenchik I, Dardick I, Auger M (1996). "Cytopathology and ultrastructure of primary rhabdoid tumor of lung". Ultrastructural Pathology. 20 (4): 355–360. doi:10.3109/01913129609016337. PMID8837343.
↑ Roggli VL, Vollmer RT, Greenberg SD, McGavran MH, Spjut HJ, Yesner R (June 1985). "Lung cancer heterogeneity: a blinded and randomized study of 100 consecutive cases". Human Pathology. 16 (6): 569–579. doi:10.1016/S0046-8177(85)80106-4. PMID2987102.
1 2 3 Rossi G, Marchioni A, Sartori G, Longo L, Piccinini S, Cavazza A (2007). "Histotype in non-small cell lung cancer therapy and staging: The emerging role of an old and underrated factor". Curr Respir Med Rev. 3: 69–77. doi:10.2174/157339807779941820. S2CID52904357.
1 2 3 Pardo J, Martinez-Peñuela AM, Sola JJ, Panizo A, Gúrpide A, Martinez-Peñuela JM, Lozano MD (October 2009). "Large cell carcinoma of the lung: an endangered species?". Applied Immunohistochemistry & Molecular Morphology. 17 (5): 383–392. doi:10.1097/PAI.0b013e31819bfd59. PMID19444077. S2CID24944728.
1 2 3 Miyagi J, Tsuhako K, Kinjo T, Iwamasa T, Hashimoto H, Ishikawa S (July 2000). "Rhabdoid tumour of the lung is a dedifferentiated phenotype of pulmonary adenocarcinoma". Histopathology. 37 (1): 37–44. doi:10.1046/j.1365-2559.2000.00906.x. PMID10931217. S2CID33018365.
1 2 Attems JH, Lintner F (2001). "Pseudomesotheliomatous adenocarcinoma of the lung with rhabdoid features". Pathology, Research and Practice. 197 (12): 841–846. doi:10.1078/0344-0338-00170. PMID11795833.
1 2 Hasegawa S, Suda T, Negi K, Hattori Y (January 2007). "Lung large cell carcinoma producing granulocyte-colony-stimulating factor". The Annals of Thoracic Surgery. 83 (1): 308–310. doi:10.1016/j.athoracsur.2006.04.049. PMID17184692.
↑ Vogel AM, Gown AM, Caughlan J, Haas JE, Beckwith JB (February 1984). "Rhabdoid tumors of the kidney contain mesenchymal specific and epithelial specific intermediate filament proteins". Laboratory Investigation; A Journal of Technical Methods and Pathology. 50 (2): 232–238. PMID6198560.
↑ Moll R, Franke WW, Schiller DL, Geiger B, Krepler R (November 1982). "The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells". Cell. 31 (1): 11–24. doi:10.1016/0092-8674(82)90400-7. PMID6186379. S2CID33701361.
↑ Song DE, Jang SJ, Black J, Ro JY (September 2007). "Mucinous bronchioloalveolar carcinoma of lung with a rhabdoid component--report of a case and review of the literature". Histopathology. 51 (3): 427–430. doi:10.1111/j.1365-2559.2007.02784.x. PMID17727492. S2CID29727857.
↑ Zinner RG, Novello S, Peng G, Herbst R, Obasaju C, Scagliotti G (March 2010). "Comparison of patient outcomes according to histology among pemetrexed-treated patients with stage IIIB/IV non-small-cell lung cancer in two phase II trials". Clinical Lung Cancer. 11 (2): 126–131. doi:10.3816/CLC.2010.n.017. PMID20199979.
↑ Reck M, von Pawel J, Zatloukal P, Ramlau R, Gorbounova V, Hirsh V, etal. (March 2009). "Phase III trial of cisplatin plus gemcitabine with either placebo or bevacizumab as first-line therapy for nonsquamous non-small-cell lung cancer: AVAil". Journal of Clinical Oncology. 27 (8): 1227–1234. doi:10.1200/JCO.2007.14.5466. PMID19188680.
↑ Chetty R, Bhana B, Batitang S, Govender D (October 1997). "Lung carcinomas composed of rhabdoid cells". European Journal of Surgical Oncology. 23 (5): 432–434. doi:10.1016/S0748-7983(97)93725-2. PMID9393573.
↑ Sheikh SS, Al-Khatti AA, Amr SS (February 2008). "Metachronus malignant rhabdoid tumor of the ileum and adenocarcinoma of lung: a unique case report". Annals of Diagnostic Pathology. 12 (1): 57–61. doi:10.1016/j.anndiagpath.2006.06.007. PMID18164418.
↑ Colby TV, Koss MN, Travis WD (1995). "Carcinoid and other neuroendocrine tumors". In Travis WD (ed.). Tumors of Lower Respiratory Tract. Washington DC: Armed Forces Institute of Pathology. p.311.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.