
A tablet is a pharmaceutical oral dosage form or solid unit dosage form. Tablets may be defined as the solid unit dosage form of medication with suitable excipients. It comprises a mixture of active substances and excipients, usually in powder form, that are pressed or compacted into a solid dose. The main advantages of tablets are that they ensure a consistent dose of medicine that is easy to consume.

In pharmacology and toxicology, a route of administration is the way by which a drug, fluid, poison, or other substance is taken into the body.
In pharmacology, bioavailability is a subcategory of absorption and is the fraction (%) of an administered drug that reaches the systemic circulation.
In the physical sciences, a partition coefficient (P) or distribution coefficient (D) is the ratio of concentrations of a compound in a mixture of two immiscible solvents at equilibrium. This ratio is therefore a comparison of the solubilities of the solute in these two liquids. The partition coefficient generally refers to the concentration ratio of un-ionized species of compound, whereas the distribution coefficient refers to the concentration ratio of all species of the compound.

ADME is an abbreviation in pharmacokinetics and pharmacology for "absorption, distribution, metabolism, and excretion", and describes the disposition of a pharmaceutical compound within an organism. The four criteria all influence the drug levels and kinetics of drug exposure to the tissues and hence influence the performance and pharmacological activity of the compound as a drug. Sometimes, liberation and/or toxicity are also considered, yielding LADME, ADMET, or LADMET.
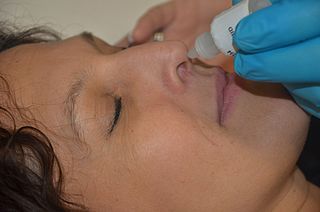
A topical medication is a medication that is applied to a particular place on or in the body. Most often topical medication means application to body surfaces such as the skin or mucous membranes to treat ailments via a large range of classes including creams, foams, gels, lotions, and ointments. Many topical medications are epicutaneous, meaning that they are applied directly to the skin. Topical medications may also be inhalational, such as asthma medications, or applied to the surface of tissues other than the skin, such as eye drops applied to the conjunctiva, or ear drops placed in the ear, or medications applied to the surface of a tooth. The word topical derives from Greek τοπικόςtopikos, "of a place".
An excipient is a substance formulated alongside the active ingredient of a medication, included for the purpose of long-term stabilization, bulking up solid formulations that contain potent active ingredients in small amounts, or to confer a therapeutic enhancement on the active ingredient in the final dosage form, such as facilitating drug absorption, reducing viscosity, or enhancing solubility. Excipients can also be useful in the manufacturing process, to aid in the handling of the active substance concerns such as by facilitating powder flowability or non-stick properties, in addition to aiding in vitro stability such as prevention of denaturation or aggregation over the expected shelf life. The selection of appropriate excipients also depends upon the route of administration and the dosage form, as well as the active ingredient and other factors. A comprehensive classification system based on structure-property-application relationships has been proposed for excipients used in parenteral medications.

Bioequivalence is a term in pharmacokinetics used to assess the expected in vivo biological equivalence of two proprietary preparations of a drug. If two products are said to be bioequivalent it means that they would be expected to be, for all intents and purposes, the same.
Drug interactions occur when a drug's mechanism of action is affected by the concomitant administration of substances such as foods, beverages, or other drugs. The cause is often inhibition of, or less effective action, of the specific receptors available to the drug. This influences drug molecules to bind to secondary targets, which may result in an array of unwanted side-effects.

Physiologically based pharmacokinetic (PBPK) modeling is a mathematical modeling technique for predicting the absorption, distribution, metabolism and excretion (ADME) of synthetic or natural chemical substances in humans and other animal species. PBPK modeling is used in pharmaceutical research and drug development, and in health risk assessment for cosmetics or general chemicals.
In pharmacology, clearance is a pharmacokinetic measurement of the volume of plasma from which a substance is completely removed per unit time. Usually, clearance is measured in L/h or mL/min. The quantity reflects the rate of drug elimination divided by plasma concentration. Excretion, on the other hand, is a measurement of the amount of a substance removed from the body per unit time. While clearance and excretion of a substance are related, they are not the same thing. The concept of clearance was described by Thomas Addis, a graduate of the University of Edinburgh Medical School.
Biological half-life is the time taken for concentration of a biological substance to decrease from its maximum concentration (Cmax) to half of Cmax in the blood plasma. It is denoted by the abbreviation .
Distribution in pharmacology is a branch of pharmacokinetics which describes the reversible transfer of a drug from one location to another within the body.
Absorption is the journey of a drug travelling from the site of administration to the site of action.
Pharmaceutical formulation, in pharmaceutics, is the process in which different chemical substances, including the active drug, are combined to produce a final medicinal product. The word formulation is often used in a way that includes dosage form.
Modified-release dosage is a mechanism that delivers a drug with a delay after its administration or for a prolonged period of time or to a specific target in the body.
Pharmacokinetics, sometimes abbreviated as PK, is a branch of pharmacology dedicated to determining the fate of substances administered to a living organism. The substances of interest include any chemical xenobiotic such as: pharmaceutical drugs, pesticides, food additives, cosmetics, etc. It attempts to analyze chemical metabolism and to discover the fate of a chemical from the moment that it is administered up to the point at which it is completely eliminated from the body. Pharmacokinetics is the study of how an organism affects a drug, whereas pharmacodynamics (PD) is the study of how the drug affects the organism. Both together influence dosing, benefit, and adverse effects, as seen in PK/PD models.
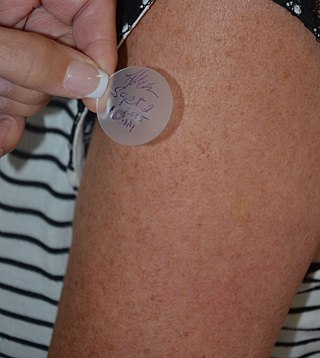
Transdermal is a route of administration wherein active ingredients are delivered across the skin for systemic distribution. Examples include transdermal patches used for medicine delivery. The drug is administered in the form of a patch or ointment that delivers the drug into the circulation for systemic effect.
Cmin is a term used in pharmacokinetics for the minimum blood plasma concentration reached by a drug during a dosing interval, which is the time interval between administration of two doses. This definition is slightly different from Ctrough, the concentration immediately prior to administration of the next dose. Cmin is the opposite of Cmax, the maximum concentration that the drug reaches. Cmin must be above certain thresholds, such as the minimum inhibitory concentration (MIC), to achieve a therapeutic effect.

In pharmacology the elimination or excretion of a drug is understood to be any one of a number of processes by which a drug is eliminated from an organism either in an unaltered form or modified as a metabolite. The kidney is the main excretory organ although others exist such as the liver, the skin, the lungs or glandular structures, such as the salivary glands and the lacrimal glands. These organs or structures use specific routes to expel a drug from the body, these are termed elimination pathways:


