A base pair (bp) is a fundamental unit of double-stranded nucleic acids consisting of two nucleobases bound to each other by hydrogen bonds. They form the building blocks of the DNA double helix and contribute to the folded structure of both DNA and RNA. Dictated by specific hydrogen bonding patterns, "Watson–Crick" base pairs allow the DNA helix to maintain a regular helical structure that is subtly dependent on its nucleotide sequence. The complementary nature of this based-paired structure provides a redundant copy of the genetic information encoded within each strand of DNA. The regular structure and data redundancy provided by the DNA double helix make DNA well suited to the storage of genetic information, while base-pairing between DNA and incoming nucleotides provides the mechanism through which DNA polymerase replicates DNA and RNA polymerase transcribes DNA into RNA. Many DNA-binding proteins can recognize specific base-pairing patterns that identify particular regulatory regions of genes.

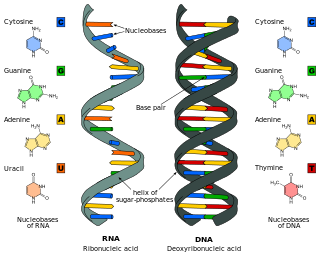
Deoxyribonucleic acid is a polymer composed of two polynucleotide chains that coil around each other to form a double helix. The polymer carries genetic instructions for the development, functioning, growth and reproduction of all known organisms and many viruses. DNA and ribonucleic acid (RNA) are nucleic acids. Alongside proteins, lipids and complex carbohydrates (polysaccharides), nucleic acids are one of the four major types of macromolecules that are essential for all known forms of life.
Nucleic acids are biopolymers, macromolecules, essential to all known forms of life. They are composed of nucleotides, which are the monomers made of three components: a 5-carbon sugar, a phosphate group and a nitrogenous base. The two main classes of nucleic acids are deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). If the sugar is ribose, the polymer is RNA; if the sugar is the ribose derivative deoxyribose, the polymer is DNA.

Nucleotides are organic molecules consisting of a nucleoside and a phosphate. They serve as monomeric units of the nucleic acid polymers – deoxyribonucleic acid (DNA) and ribonucleic acid (RNA), both of which are essential biomolecules within all life-forms on Earth. Nucleotides are obtained in the diet and are also synthesized from common nutrients by the liver.
Ribonucleic acid (RNA) is a polymeric molecule essential in various biological roles in coding, decoding, regulation and expression of genes. RNA and deoxyribonucleic acid (DNA) are nucleic acids. Along with lipids, proteins, and carbohydrates, nucleic acids constitute one of the four major macromolecules essential for all known forms of life. Like DNA, RNA is assembled as a chain of nucleotides, but unlike DNA, RNA is found in nature as a single strand folded onto itself, rather than a paired double strand. Cellular organisms use messenger RNA (mRNA) to convey genetic information that directs synthesis of specific proteins. Many viruses encode their genetic information using an RNA genome.
The RNA world is a hypothetical stage in the evolutionary history of life on Earth, in which self-replicating RNA molecules proliferated before the evolution of DNA and proteins. The term also refers to the hypothesis that posits the existence of this stage.

Nucleobases, also known as nitrogenous bases or often simply bases, are nitrogen-containing biological compounds that form nucleosides, which, in turn, are components of nucleotides, with all of these monomers constituting the basic building blocks of nucleic acids. The ability of nucleobases to form base pairs and to stack one upon another leads directly to long-chain helical structures such as ribonucleic acid (RNA) and deoxyribonucleic acid (DNA). Five nucleobases—adenine (A), cytosine (C), guanine (G), thymine (T), and uracil (U)—are called primary or canonical. They function as the fundamental units of the genetic code, with the bases A, G, C, and T being found in DNA while A, G, C, and U are found in RNA. Thymine and uracil are distinguished by merely the presence or absence of a methyl group on the fifth carbon (C5) of these heterocyclic six-membered rings. In addition, some viruses have aminoadenine (Z) instead of adenine. It differs in having an extra amine group, creating a more stable bond to thymine.
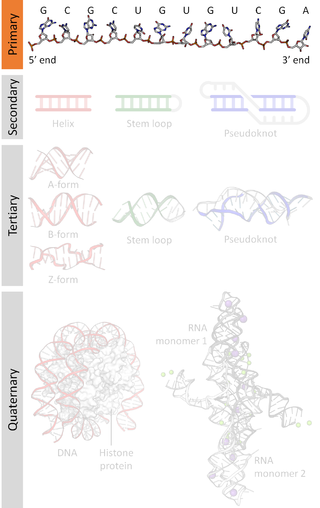
A nucleic acid sequence is a succession of bases signified by a series of a set of five different letters that indicate the order of nucleotides forming alleles within a DNA or RNA (GACU) molecule. By convention, sequences are usually presented from the 5' end to the 3' end. For DNA, the sense strand is used. Because nucleic acids are normally linear (unbranched) polymers, specifying the sequence is equivalent to defining the covalent structure of the entire molecule. For this reason, the nucleic acid sequence is also termed the primary structure.

In biochemistry, a ribonucleotide is a nucleotide containing ribose as its pentose component. It is considered a molecular precursor of nucleic acids. Nucleotides are the basic building blocks of DNA and RNA. Ribonucleotides themselves are basic monomeric building blocks for RNA. Deoxyribonucleotides, formed by reducing ribonucleotides with the enzyme ribonucleotide reductase (RNR), are essential building blocks for DNA. There are several differences between DNA deoxyribonucleotides and RNA ribonucleotides. Successive nucleotides are linked together via phosphodiester bonds.
The history of molecular biology begins in the 1930s with the convergence of various, previously distinct biological and physical disciplines: biochemistry, genetics, microbiology, virology and physics. With the hope of understanding life at its most fundamental level, numerous physicists and chemists also took an interest in what would become molecular biology.

In molecular biology, G-quadruplex secondary structures (G4) are formed in nucleic acids by sequences that are rich in guanine. They are helical in shape and contain guanine tetrads that can form from one, two or four strands. The unimolecular forms often occur naturally near the ends of the chromosomes, better known as the telomeric regions, and in transcriptional regulatory regions of multiple genes, both in microbes and across vertebrates including oncogenes in humans. Four guanine bases can associate through Hoogsteen hydrogen bonding to form a square planar structure called a guanine tetrad, and two or more guanine tetrads can stack on top of each other to form a G-quadruplex.

Biomolecular structure is the intricate folded, three-dimensional shape that is formed by a molecule of protein, DNA, or RNA, and that is important to its function. The structure of these molecules may be considered at any of several length scales ranging from the level of individual atoms to the relationships among entire protein subunits. This useful distinction among scales is often expressed as a decomposition of molecular structure into four levels: primary, secondary, tertiary, and quaternary. The scaffold for this multiscale organization of the molecule arises at the secondary level, where the fundamental structural elements are the molecule's various hydrogen bonds. This leads to several recognizable domains of protein structure and nucleic acid structure, including such secondary-structure features as alpha helixes and beta sheets for proteins, and hairpin loops, bulges, and internal loops for nucleic acids. The terms primary, secondary, tertiary, and quaternary structure were introduced by Kaj Ulrik Linderstrøm-Lang in his 1951 Lane Medical Lectures at Stanford University.
Nucleic acid thermodynamics is the study of how temperature affects the nucleic acid structure of double-stranded DNA (dsDNA). The melting temperature (Tm) is defined as the temperature at which half of the DNA strands are in the random coil or single-stranded (ssDNA) state. Tm depends on the length of the DNA molecule and its specific nucleotide sequence. DNA, when in a state where its two strands are dissociated, is referred to as having been denatured by the high temperature.
Nucleic acid analogues are compounds which are analogous to naturally occurring RNA and DNA, used in medicine and in molecular biology research. Nucleic acids are chains of nucleotides, which are composed of three parts: a phosphate backbone, a pentose sugar, either ribose or deoxyribose, and one of four nucleobases. An analogue may have any of these altered. Typically the analogue nucleobases confer, among other things, different base pairing and base stacking properties. Examples include universal bases, which can pair with all four canonical bases, and phosphate-sugar backbone analogues such as PNA, which affect the properties of the chain . Nucleic acid analogues are also called Xeno Nucleic Acid and represent one of the main pillars of xenobiology, the design of new-to-nature forms of life based on alternative biochemistries.

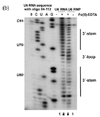
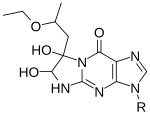
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide is a water-soluble carbodiimide usually handled as the hydrochloride. It is typically employed in the 4.0-6.0 pH range. It is generally used as a carboxyl activating agent for the coupling of primary amines to yield amide bonds. While other carbodiimides like dicyclohexylcarbodiimide (DCC) or diisopropylcarbodiimide (DIC) are also employed for this purpose, EDC has the advantage that the urea byproduct formed can be washed away from the amide product using dilute acid. Additionally, EDC can also be used to activate phosphate groups in order to form phosphomonoesters and phosphodiesters. Common uses for this carbodiimide include peptide synthesis, protein crosslinking to nucleic acids, but also in the preparation of immunoconjugates. EDC is often used in combination with N-hydroxysuccinimide (NHS) for the immobilisation of large biomolecules. Recent work has also used EDC to assess the structure state of uracil nucleobases in RNA.

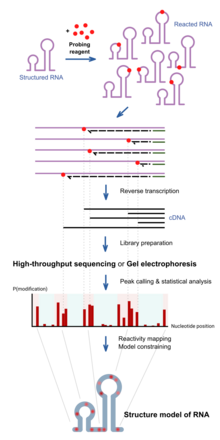
Nucleic acid secondary structure is the basepairing interactions within a single nucleic acid polymer or between two polymers. It can be represented as a list of bases which are paired in a nucleic acid molecule. The secondary structures of biological DNAs and RNAs tend to be different: biological DNA mostly exists as fully base paired double helices, while biological RNA is single stranded and often forms complex and intricate base-pairing interactions due to its increased ability to form hydrogen bonds stemming from the extra hydroxyl group in the ribose sugar.
Nucleic acid NMR is the use of nuclear magnetic resonance spectroscopy to obtain information about the structure and dynamics of nucleic acid molecules, such as DNA or RNA. It is useful for molecules of up to 100 nucleotides, and as of 2003, nearly half of all known RNA structures had been determined by NMR spectroscopy.

Xeno nucleic acids (XNA) are synthetic nucleic acid analogues that have a different sugar backbone than the natural nucleic acids DNA and RNA. As of 2011, at least six types of synthetic sugars have been shown to form nucleic acid backbones that can store and retrieve genetic information. Research is now being done to create synthetic polymerases to transform XNA. The study of its production and application has created a field known as xenobiology.

DNA base flipping, or nucleotide flipping, is a mechanism in which a single nucleotide base, or nucleobase, is rotated outside the nucleic acid double helix. This occurs when a nucleic acid-processing enzyme needs access to the base to perform work on it, such as its excision for replacement with another base during DNA repair. It was first observed in 1994 using X-ray crystallography in a methyltransferase enzyme catalyzing methylation of a cytosine base in DNA. Since then, it has been shown to be used by different enzymes in many biological processes such as DNA methylation, various DNA repair mechanisms, and DNA replication. It can also occur in RNA double helices or in the DNA:RNA intermediates formed during RNA transcription.
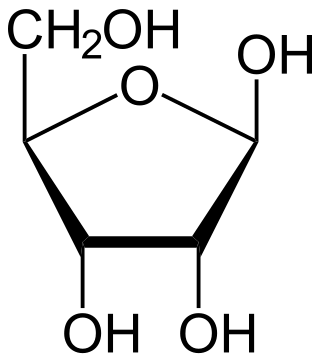
Ribose is a simple sugar and carbohydrate with molecular formula C5H10O5 and the linear-form composition H−(C=O)−(CHOH)4−H. The naturally-occurring form, d-ribose, is a component of the ribonucleotides from which RNA is built, and so this compound is necessary for coding, decoding, regulation and expression of genes. It has a structural analog, deoxyribose, which is a similarly essential component of DNA. l-ribose is an unnatural sugar that was first prepared by Emil Fischer and Oscar Piloty in 1891. It was not until 1909 that Phoebus Levene and Walter Jacobs recognised that d-ribose was a natural product, the enantiomer of Fischer and Piloty's product, and an essential component of nucleic acids. Fischer chose the name "ribose" as it is a partial rearrangement of the name of another sugar, arabinose, of which ribose is an epimer at the 2' carbon; both names also relate to gum arabic, from which arabinose was first isolated and from which they prepared l-ribose.