
In chemistry, a hydrogen bond (H-bond) is a specific type of molecular interaction that exhibits partial covalent character and cannot be described as a purely electrostatic force. It occurs when a hydrogen (H) atom, covalently bonded to a more electronegative donor atom or group (Dn), interacts with another electronegative atom bearing a lone pair of electrons—the hydrogen bond acceptor (Ac). Unlike simple dipole–dipole interactions, hydrogen bonding arises from charge transfer, orbital interactions, and quantum mechanical delocalization, making it a resonance-assisted interaction rather than a mere electrostatic attraction.
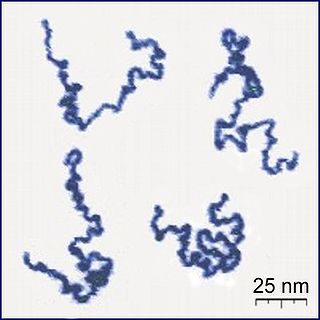
A polymer is a substance or material that consists of very large molecules, or macromolecules, that are constituted by many repeating subunits derived from one or more species of monomers. Due to their broad spectrum of properties, both synthetic and natural polymers play essential and ubiquitous roles in everyday life. Polymers range from familiar synthetic plastics such as polystyrene to natural biopolymers such as DNA and proteins that are fundamental to biological structure and function. Polymers, both natural and synthetic, are created via polymerization of many small molecules, known as monomers. Their consequently large molecular mass, relative to small molecule compounds, produces unique physical properties including toughness, high elasticity, viscoelasticity, and a tendency to form amorphous and semicrystalline structures rather than crystals.

A micelle or micella is an aggregate of surfactant amphipathic lipid molecules dispersed in a liquid, forming a colloidal suspension. A typical micelle in water forms an aggregate with the hydrophilic "head" regions in contact with surrounding solvent, sequestering the hydrophobic single-tail regions in the micelle centre.

Supramolecular chemistry refers to the branch of chemistry concerning chemical systems composed of a discrete number of molecules. The strength of the forces responsible for spatial organization of the system range from weak intermolecular forces, electrostatic charge, or hydrogen bonding to strong covalent bonding, provided that the electronic coupling strength remains small relative to the energy parameters of the component. While traditional chemistry concentrates on the covalent bond, supramolecular chemistry examines the weaker and reversible non-covalent interactions between molecules. These forces include hydrogen bonding, metal coordination, hydrophobic forces, van der Waals forces, pi–pi interactions and electrostatic effects.
A polycatenane is a chemical substance that, like polymers, is chemically constituted by a large number of units. These units are made up of concatenated rings into a chain-like structure.

In supramolecular chemistry, host–guest chemistry describes complexes that are composed of two or more molecules or ions that are held together in unique structural relationships by forces other than those of full covalent bonds. Host–guest chemistry encompasses the idea of molecular recognition and interactions through non-covalent bonding. Non-covalent bonding is critical in maintaining the 3D structure of large molecules, such as proteins and is involved in many biological processes in which large molecules bind specifically but transiently to one another.

Molecular imprinting is a technique to create template-shaped cavities in polymer matrices with predetermined selectivity and high affinity. This technique is based on the system used by enzymes for substrate recognition, which is called the "lock and key" model. The active binding site of an enzyme has a shape specific to a substrate. Substrates with a complementary shape to the binding site selectively bind to the enzyme; alternative shapes that do not fit the binding site are not recognized.
Dynamic covalent chemistry (DCvC) is a synthetic strategy employed by chemists to make complex molecular and supramolecular assemblies from discrete molecular building blocks. DCvC has allowed access to complex assemblies such as covalent organic frameworks, molecular knots, polymers, and novel macrocycles. Not to be confused with dynamic combinatorial chemistry, DCvC concerns only covalent bonding interactions. As such, it only encompasses a subset of supramolecular chemistries.

In chemistry, a salt bridge is a combination of two non-covalent interactions: hydrogen bonding and ionic bonding. Ion pairing is one of the most important noncovalent forces in chemistry, in biological systems, in different materials and in many applications such as ion pair chromatography. It is a most commonly observed contribution to the stability to the entropically unfavorable folded conformation of proteins. Although non-covalent interactions are known to be relatively weak interactions, small stabilizing interactions can add up to make an important contribution to the overall stability of a conformer. Not only are salt bridges found in proteins, but they can also be found in supramolecular chemistry. The thermodynamics of each are explored through experimental procedures to access the free energy contribution of the salt bridge to the overall free energy of the state.
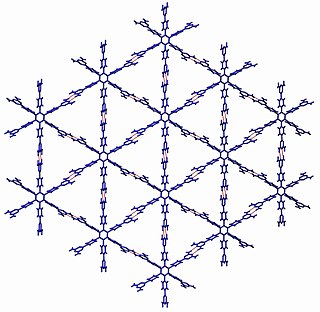
Crystal engineering studies the design and synthesis of solid-state structures with desired properties through deliberate control of intermolecular interactions. It is an interdisciplinary academic field, bridging solid-state and supramolecular chemistry.

Self-healing materials are artificial or synthetically created substances that have the built-in ability to automatically repair damages to themselves without any external diagnosis of the problem or human intervention. Generally, materials will degrade over time due to fatigue, environmental conditions, or damage incurred during operation. Cracks and other types of damage on a microscopic level have been shown to change thermal, electrical, and acoustical properties of materials, and the propagation of cracks can lead to eventual failure of the material. In general, cracks are hard to detect at an early stage, and manual intervention is required for periodic inspections and repairs. In contrast, self-healing materials counter degradation through the initiation of a repair mechanism that responds to the micro-damage. Some self-healing materials are classed as smart structures, and can adapt to various environmental conditions according to their sensing and actuation properties.
In chemistry, a halogen bond occurs when there is evidence of a net attractive interaction between an electrophilic region associated with a halogen atom in a molecular entity and a nucleophilic region in another, or the same, molecular entity. Like a hydrogen bond, the result is not a formal chemical bond, but rather a strong electrostatic attraction. Mathematically, the interaction can be decomposed in two terms: one describing an electrostatic, orbital-mixing charge-transfer and another describing electron-cloud dispersion. Halogen bonds find application in supramolecular chemistry; drug design and biochemistry; crystal engineering and liquid crystals; and organic catalysis.

Takuzo Aida is a polymer chemist known for his work in the fields of supramolecular chemistry, materials chemistry and polymer chemistry. Aida, who is the Deputy Director for the RIKEN Center for Emergent Matter Science (CEMS) and a Distinguished University Professor at the University of Tokyo, has made pioneering contributions to the initiation, fundamental progress, and conceptual expansion of supramolecular polymerization. Aida has also been a leader and advocate for addressing critical environmental issues caused by plastic waste and microplastics in the oceans, soil, and food supply, through the development of dynamic, responsive, healable, reorganizable, and adaptive supramolecular polymers and related soft materials.

Egbert (Bert) Willem Meijer is a Dutch organic chemist, known for his work in the fields of supramolecular chemistry, materials chemistry and polymer chemistry. Meijer, who is distinguished professor of Molecular Sciences at Eindhoven University of Technology (TU/e) and Academy Professor of the Royal Netherlands Academy of Arts and Sciences, is considered one of the founders of the field of supramolecular polymer chemistry. Meijer is a prolific author, sought-after academic lecturer and recipient of multiple awards in the fields of organic and polymer chemistry.

Peptide amphiphiles (PAs) are peptide-based molecules that self-assemble into supramolecular nanostructures including; spherical micelles, twisted ribbons, and high-aspect-ratio nanofibers. A peptide amphiphile typically comprises a hydrophilic peptide sequence attached to a lipid tail, i.e. a hydrophobic alkyl chain with 10 to 16 carbons. Therefore, they can be considered a type of lipopeptide. A special type of PA, is constituted by alternating charged and neutral residues, in a repeated pattern, such as RADA16-I. The PAs were developed in the 1990s and the early 2000s and could be used in various medical areas including: nanocarriers, nanodrugs, and imaging agents. However, perhaps their main potential is in regenerative medicine to culture and deliver cells and growth factors.
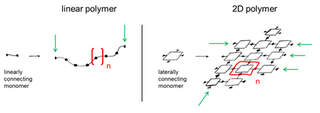
A two-dimensional polymer (2DP) is a sheet-like monomolecular macromolecule consisting of laterally connected repeat units with end groups along all edges. This recent definition of 2DP is based on Hermann Staudinger's polymer concept from the 1920s. According to this, covalent long chain molecules ("Makromoleküle") do exist and are composed of a sequence of linearly connected repeat units and end groups at both termini.
Self-healing hydrogels are a specialized type of polymer hydrogel. A hydrogel is a macromolecular polymer gel constructed of a network of crosslinked polymer chains. Hydrogels are synthesized from hydrophilic monomers by either chain or step growth, along with a functional crosslinker to promote network formation. A net-like structure along with void imperfections enhance the hydrogel's ability to absorb large amounts of water via hydrogen bonding. As a result, hydrogels, self-healing alike, develop characteristic firm yet elastic mechanical properties. Self-healing refers to the spontaneous formation of new bonds when old bonds are broken within a material. The structure of the hydrogel along with electrostatic attraction forces drive new bond formation through reconstructive covalent dangling side chain or non-covalent hydrogen bonding. These flesh-like properties have motivated the research and development of self-healing hydrogels in fields such as reconstructive tissue engineering as scaffolding, as well as use in passive and preventive applications.

Polyrotaxane is a type of mechanically interlocked molecule consisting of strings and rings, in which multiple rings are threaded onto a molecular axle and prevented from dethreading by two bulky end groups. As oligomeric or polymeric species of rotaxanes, polyrotaxanes are also capable of converting energy input to molecular movements because the ring motions can be controlled by external stimulus. Polyrotaxanes have attracted much attention for decades, because they can help build functional molecular machines with complicated molecular structure.

In host–guest chemistry, macromolecular cages are a type of macromolecule structurally consisting of a three-dimensional chamber surrounded by a molecular framework. Macromolecular cage architectures come in various sizes ranging from 1-50 nm and have varying topologies as well as functions. They can be synthesized through covalent bonding or self-assembly through non-covalent interactions. Most macromolecular cages that are formed through self-assembly are sensitive to pH, temperature, and solvent polarity.
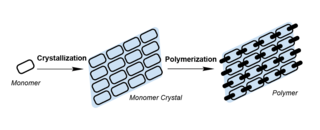
Topochemical polymerization is a polymerization method performed by monomers aligned in the crystal state. In this process, the monomers are crystallised and polymerised under external stimuli such as heat, light, or pressure. Compared to traditional polymerisation, the movement of monomers was confined by the crystal lattice in topochemical polymerisation, giving rise to polymers with high crystallinity, tacticity, and purity. Topochemical polymerisation can also be used to synthesise unique polymers such as polydiacetylene that are otherwise hard to prepare.



