Two-Dimensional Nuclear Magnetic Resonance (2D NMR) is an advanced spectroscopic technique that builds upon the capabilities of one-dimensional (1D) NMR by incorporating an additional frequency dimension. This extension allows for a more comprehensive analysis of molecular structures.[1] In 2D NMR, signals are distributed across two frequency axes, providing improved resolution and separation of overlapping peaks, particularly beneficial for studying complex molecules. This technique identifies correlations between different nuclei within a molecule, facilitating the determination of connectivity, spatial proximity, and dynamic interactions.
2D NMR encompasses a variety of experiments,[1] including COSY (Correlation Spectroscopy), TOCSY (Total Correlation Spectroscopy), NOESY (Nuclear Overhauser Effect Spectroscopy), and HSQC (Heteronuclear Single Quantum Coherence). These techniques are indispensable in fields such as structural biology, where they are pivotal in determining protein and nucleic acid structures; organic chemistry, where they aid in elucidating complex organic molecules; and materials science, where they offer insights into molecular interactions in polymers and metal-organic frameworks. By resolving signals that would typically overlap in the 1D NMR spectra of complex molecules, 2D NMR enhances the clarity of structural information.[1] 2D NMR can provide detailed information about the chemical structure and the three-dimensional arrangement of molecules.
The first two-dimensional experiment, COSY, was proposed by Jean Jeener, a professor at the Université Libre de Bruxelles, in 1971. This experiment was later implemented by Walter P. Aue, Enrico Bartholdi and Richard R. Ernst, who published their work in 1976.[2][3][4]
Fundamental concepts
Each experiment consists of a sequence of radio frequency (RF) pulses with delay periods in between them. The timing, frequencies, and intensities of these pulses distinguish different NMR experiments from one another.[5] Almost all two-dimensional experiments have four stages: the preparation period, where a magnetization coherence is created through a set of RF pulses; the evolution period, a determined length of time during which no pulses are delivered and the nuclear spins are allowed to freely precess (rotate); the mixing period, where the coherence is manipulated by another series of pulses into a state which will give an observable signal; and the detection period, in which the free induction decay signal from the sample is observed as a function of time, in a manner identical to one-dimensional FT-NMR.[6]
The two dimensions of a two-dimensional NMR experiment are two frequency axes representing a chemical shift. Each frequency axis is associated with one of the two time variables, which are the length of the evolution period (the evolution time) and the time elapsed during the detection period (the detection time). They are each converted from a time series to a frequency series through a two-dimensional Fourier transform. A single two-dimensional experiment is generated as a series of one-dimensional experiments, with a different specific evolution time in successive experiments, with the entire duration of the detection period recorded in each experiment.[6]
The end result is a plot showing an intensity value for each pair of frequency variables. The intensities of the peaks in the spectrum can be represented using a third dimension. More commonly, intensity is indicated using contour lines or different colors.
Homonuclear through-bond correlation methods
In these methods, magnetization transfer occurs between nuclei of the same type, through J-coupling of nuclei connected by up to a few bonds.
Correlation spectroscopy (COSY)
In standard COSY, the preparation (p1) and mixing (p2) periods each consist of a single 90° pulse separated by the evolution time t1, and the resonance signal from the sample is read during the detection period over a range of times t2.
The first and most popular two-dimension NMR experiment is the homonuclear correlation spectroscopy (COSY) sequence, which is used to identify spins which are coupled to each other. It consists of a single RF pulse (p1) followed by the specific evolution time (t1) followed by a second pulse (p2) followed by a measurement period (t2).[7]
The Correlation Spectroscopy experiment operates by correlating nuclei coupled to each other through scalar coupling, also known as J-coupling.[8] This coupling is the interaction between nuclear spins connected by bonds, typically observed between nuclei that are 2-3 bonds apart (e.g., vicinal protons). By detecting these interactions, COSY provides vital information about the connectivity between atoms within a molecule, making it a crucial tool for structural elucidation in organic chemistry.
The COSY experiment generates a two-dimensional spectrum with chemical shifts along the x-axis (horizontal) and y-axis (vertical) and involves several key steps.[1] First, the sample is excited using a series of radiofrequency (RF) pulses, bringing the nuclear spins into a higher energy state. After the first RF pulse, the system evolves freely for a period called t1, during which the spins precess at frequencies corresponding to their chemical shifts. The correlation between nuclei is achieved by incrementally varying the evolution time (t1) to capture indirect interactions. This series of experiments, each with a different value of t1, allows for the detection of chemical shifts from nuclei that may not be observed directly in a one-dimensional spectrum. As t1 is incremented, cross-peaks are produced in the resulting 2D spectrum, representing interactions like coupling or spatial proximity between nuclei. This approach helps map out atomic connections, providing deeper insight into molecular structure and aiding in the interpretation of complex systems.
Cross peaks result from a phenomenon called magnetization transfer, and their presence indicates that two nuclei are coupled which have the two different chemical shifts that make up the cross peak's coordinates. Each coupling gives two symmetrical cross peaks above and below the diagonal. That is, a cross-peak occurs when there is a correlation between the signals of the spectrum along each of the two axes at these values. An easy visual way to determine which couplings a cross peak represents is to find the diagonal peak which is directly above or below the cross peak, and the other diagonal peak which is directly to the left or right of the cross peak. The nuclei represented by those two diagonal peaks are coupled.[7]
Next, a second RF pulse is applied to allow magnetization to transfer between coupled nuclei. The resulting signal is recorded continuously during a detection period ( t2) after the second RF pulse. The data are then processed through Fourier transformation along both the t1 and t2 axes, creating a 2D spectrum with peaks plotted along the diagonal and off-diagonal.
When interpreting the COSY spectrum, diagonal peaks correspond to the 1D chemical shifts of individual nuclei, similar to the standard peaks in a 1D NMR spectrum. The key feature of a COSY spectrum is the presence of cross-peaks as shown in Figure 1, indicating coupling between pairs of nuclei. These cross-peaks provide crucial information about the connectivity within a molecule, showing that the two nuclei are connected by a small number of bonds, usually two or three bonds.
COSY is especially useful when dealing with complex molecules such as natural products, peptides, and proteins, where understanding the connectivity of different nuclei through bonds is crucial. While 1D NMR is more straightforward and ideal for identifying basic structural features, COSY enhances the capabilities of NMR by providing deeper insights into molecular connectivity.
The two-dimensional spectrum that results from the COSY experiment shows the frequencies for a single isotope, most commonly hydrogen (1H) along both axes. (Techniques have also been devised for generating heteronuclear correlation spectra, in which the two axes correspond to different isotopes, such as 13C and 1H.) Diagonal peaks correspond to the peaks in a 1D-NMR experiment, while the cross peaks indicate couplings between pairs of nuclei (much as multiplet splitting indicates couplings in 1D-NMR).[7]
H COSY spectrum of progesterone. The spectrum that appears along both the horizontal and vertical axes is a regular one dimensional H NMR spectrum. The bulk of the peaks appear along the diagonal, while cross-peaks appear symmetrically above and below the diagonal.
COSY-90 is the most common COSY experiment. In COSY-90, the p1 pulse tilts the nuclear spin by 90°. Another member of the COSY family is COSY-45. In COSY-45 a 45° pulse is used instead of a 90° pulse for the second pulse, p2. The advantage of a COSY-45 is that the diagonal-peaks are less pronounced, making it simpler to match cross-peaks near the diagonal in a large molecule. Additionally, the relative signs of the coupling constants (see J-coupling#Magnitude of J-coupling) can be elucidated from a COSY-45 spectrum. This is not possible using COSY-90.[9] Overall, the COSY-45 offers a cleaner spectrum while the COSY-90 is more sensitive.
Another related COSY technique is double quantum filtered (DQF) COSY. DQF COSY uses a coherence selection method such as phase cycling or pulsed field gradients, which cause only signals from double-quantum coherences to give an observable signal. This has the effect of decreasing the intensity of the diagonal peaks and changing their lineshape from a broad "dispersion" lineshape to a sharper "absorption" lineshape. It also eliminates diagonal peaks from uncoupled nuclei. These all have the advantage that they give a cleaner spectrum in which the diagonal peaks are prevented from obscuring the cross peaks, which are weaker in a regular COSY spectrum.[10]
The TOCSY experiment is similar to the COSY experiment, in that cross peaks of coupled protons are observed. However, cross peaks are observed not only for nuclei which are directly coupled, but also between nuclei which are connected by a chain of couplings. This makes it useful for identifying the larger interconnected networks of spin couplings. This ability is achieved by inserting a repetitive series of pulses which cause isotropic mixing during the mixing period. Longer isotropic mixing times cause the polarization to spread out through an increasing number of bonds.[11]
In the case of oligosaccharides, each sugar residue is an isolated spin system, so it is possible to differentiate all the protons of a specific sugar residue. A 1D version of TOCSY is also available, and by irradiating a single proton the rest of the spin system can be revealed. Recent advances in this technique include the 1D-CSSF (chemical shift selective filter) TOCSY experiment, which produces higher quality spectra and allows coupling constants to be reliably extracted and used to help determine stereochemistry.
TOCSY is sometimes called "homonuclear Hartmann–Hahn spectroscopy" (HOHAHA).[12]
Incredible natural-abundance double-quantum transfer experiment (INADEQUATE)
INADEQUATE[13] is a method often used to find 13C couplings between adjacent carbon atoms. Because the natural abundance of 13C is only about 1%, only about 0.01% of molecules being studied will have the two nearby 13C atoms needed for a signal in this experiment. However, correlation selection methods are used (similarly to DQF COSY) to prevent signals from single 13C atoms, so that the double 13C signals can be easily resolved. Each coupled pair of nuclei gives a pair of peaks on the INADEQUATE spectrum which both have the same vertical coordinate, which is the sum of the chemical shifts of the nuclei; the horizontal coordinate of each peak is the chemical shift for each of the nuclei separately.[14]
Heteronuclear through-bond correlation methods
Heteronuclear correlation spectroscopy gives signal based upon coupling between nuclei of two different types. Often the two nuclei are protons and another nucleus (called a "heteronucleus"). For historical reasons, experiments which record the proton rather than the heteronucleus spectrum during the detection period are called "inverse" experiments. This is because the low natural abundance of most heteronuclei would result in the proton spectrum being overwhelmed with signals from molecules with no active heteronuclei, making it useless for observing the desired, coupled signals. With the advent of techniques for suppressing these undesired signals, inverse correlation experiments such as HSQC, HMQC, and HMBC are actually much more common today. "Normal" heteronuclear correlation spectroscopy, in which the heteronucleus spectrum is recorded, is known as HETCOR.[15]
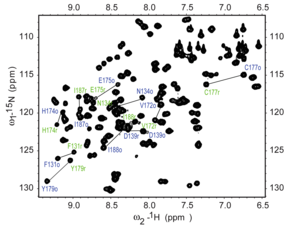
H– N HSQC spectrum of a fragment of the protein NleG3-2. Each peak in the spectrum represents a bonded N–H pair, with its two coordinates corresponding to the chemical shifts of each of the H and N atoms. Some of the peaks are labeled with the amino acid residue that gives that signal.
Heteronuclear Single Quantum Coherence (HSQC) is a 2D NMR technique utilized for the detection of interactions between different types of nuclei which are separated by one bond, particularly a proton (1H) and a heteronucleus such as carbon (13C) or nitrogen (15N).[17] This method gives one peak per pair of coupled nuclei, whose two coordinates are the chemical shifts of the two coupled atoms.[18]
This method plays a role in structural elucidation, particularly in analyzing organic compounds, natural products, and biomolecules such as proteins and nucleic acids. HSQC is designed to detect one-bond correlations between protons and heteronuclear atoms, providing insight into the connectivity of hydrogen and heteronuclear atoms through the transfer of magnetization.
The HSQC experiment involves a series of steps to generate a two-dimensional NMR spectrum. Initially, the sample is excited using radiofrequency (RF) pulses, bringing the nuclear spins into an excited state and preparing them for magnetization transfer. Magnetization is then transferred from the proton to the heteronucleus through a one-bond scalar coupling (J-coupling), ensuring that only directly bonded nuclei participate in the transfer. Subsequently, the system evolves during a period called t1, and the magnetization is transferred back from the heteronuclear to the proton. The final signal is detected, encoding both the proton and the heteronuclear information, and a Fourier transformation is performed to create a 2D spectrum correlating the proton and heteronuclear chemical shifts.
HSQC works by transferring magnetization from the I nucleus (usually the proton) to the S nucleus (usually the heteroatom) using the INEPT pulse sequence; this first step is done because the proton has a greater equilibrium magnetization and thus this step creates a stronger signal. The magnetization then evolves and then is transferred back to the I nucleus for observation. An extra spin echo step can then optionally be used to decouple the signal, simplifying the spectrum by collapsing multiplets to a single peak. The undesired uncoupled signals are removed by running the experiment twice with the phase of one specific pulse reversed; this reverses the signs of the desired but not the undesired peaks, so subtracting the two spectra will give only the desired peaks.[18]
Interpretation of the HSQC spectrum is based on the observation of cross-peaks, which indicates the direct bonding between protons and carbons or nitrogens. Each cross-peak corresponds to a specific 1H-13C or 1H-15N pair, providing direct assignments of 1H-Xconnectivity, where X is the heteronucleus[17] The HSQC technique offers several advantages, including its focus on one-bond correlations, increased sensitivity due to the direct detection of protons, and the simplification of crowded spectra by resolving overlapping signals and aiding in the analysis of complex molecules.
Heteronuclear multiple-quantum correlation spectroscopy (HMQC) gives an identical spectrum as HSQC, but using a different method. The two methods give similar quality results for small to medium-sized molecules, but HSQC is considered to be superior for larger molecules.[18]
HMBC detects heteronuclear correlations over longer ranges of about 2–4 bonds. The difficulty of detecting multiple-bond correlations is that the HSQC and HMQC sequences contain a specific delay time between pulses which allows detection only of a range around a specific coupling constant. This is not a problem for the single-bond methods since the coupling constants tend to lie in a narrow range, but multiple-bond coupling constants cover a much wider range and cannot all be captured in a single HSQC or HMQC experiment.[19]
In HMBC, this difficulty is overcome by omitting one of these delays from an HMQC sequence. This increases the range of coupling constants that can be detected, and also reduces signal loss from relaxation. The cost is that this eliminates the possibility of decoupling the spectrum, and introduces phase distortions into the signal. There is a modification of the HMBC method which suppresses one-bond signals, leaving only the multiple-bond signals.[19]
Through-space correlation methods
These methods establish correlations between nuclei which are physically close to each other regardless of whether there is a bond between them. They use the nuclear Overhauser effect (NOE) by which nearby atoms (within about 5 Å) undergo cross relaxation by a mechanism related to spin–lattice relaxation.
Nuclear Overhauser effect spectroscopy (NOESY)
In NOESY, the nuclear Overhauser cross relaxation between nuclear spins during the mixing period is used to establish the correlations. The spectrum obtained is similar to COSY, with diagonal peaks and cross peaks, however the cross peaks connect resonances from nuclei that are spatially close rather than those that are through-bond coupled to each other. NOESY spectra also contain extra axial peaks which do not provide extra information and can be eliminated through a different experiment by reversing the phase of the first pulse.[20]
One application of NOESY is in the study of large biomolecules, such as in protein NMR, in which relationships can often be assigned using sequential walking.
The NOESY experiment can also be performed in a one-dimensional fashion by pre-selecting individual resonances. The spectra are read with the pre-selected nuclei giving a large, negative signal while neighboring nuclei are identified by weaker, positive signals. This only reveals which peaks have measurable NOEs to the resonance of interest but takes much less time than the full 2D experiment. In addition, if a pre-selected nucleus changes environment within the time scale of the experiment, multiple negative signals may be observed. This offers exchange information similar to the EXSY (exchange spectroscopy) NMR method.
NOESY experiments are important tool to identify stereochemistry of a molecule in solvent whereas single crystal XRD used to identify stereochemistry of a molecule in solid form.
In HOESY, much like NOESY is used for the cross relaxation between nuclear spins. However, HOESY can offer information about other NMR active nuclei in a spatially relevant manner. Examples include any nuclei X{Y} or X→Y such as 1H→13C, 19F→13C, 31P→13C, or 77Se→13C. The experiments typically observe NOEs from protons on X, X{1H}, but do not have to include protons.[21]
ROESY is similar to NOESY, except that the initial state is different. Instead of observing cross relaxation from an initial state of z-magnetization, the equilibrium magnetization is rotated onto the x axis and then spin-locked by an external magnetic field so that it cannot precess. This method is useful for certain molecules whose rotational correlation time falls in a range where the nuclear Overhauser effect is too weak to be detectable, usually molecules with a molecular weight around 1000 daltons, because ROESY has a different dependence between the correlation time and the cross-relaxation rate constant. In NOESY the cross-relaxation rate constant goes from positive to negative as the correlation time increases, giving a range where it is near zero, whereas in ROESY the cross-relaxation rate constant is always positive.[22][23]
ROESY is sometimes called "cross relaxation appropriate for minimolecules emulated by locked spins" (CAMELSPIN).[23]
Resolved-spectrum methods
Unlike correlated spectra, resolved spectra spread the peaks in a 1D-NMR experiment into two dimensions without adding any extra peaks. These methods are usually called J-resolved spectroscopy, but are sometimes also known as chemical shift resolved spectroscopy or δ-resolved spectroscopy. They are useful for analysing molecules for which the 1D-NMR spectra contain overlapping multiplets as the J-resolved spectrum vertically displaces the multiplet from each nucleus by a different amount. Each peak in the 2D spectrum will have the same horizontal coordinate that it has in a non-decoupled 1D spectrum, but its vertical coordinate will be the chemical shift of the single peak that the nucleus has in a decoupled 1D spectrum.[24]
For the heteronuclear version, the simplest pulse sequence used is called a Müller–Kumar–Ernst (MKE) experiment, which has a single 90° pulse for the heteronucleus for the preparation period, no mixing period, and applies a decoupling signal to the proton during the detection period. There are several variants on this pulse sequence which are more sensitive and more accurate, which fall under the categories of gated decoupler methods and spin-flip methods. Homonuclear J-resolved spectroscopy uses the spin echo pulse sequence.[24]
Higher-dimensional methods
3D and 4D experiments can also be done, sometimes by running the pulse sequences from two or three 2D experiments in series. Many of the commonly used 3D experiments, however, are triple resonance experiments; examples include the HNCA and HNCOCA experiments, which are often used in protein NMR.
↑ Aue, W. P.; Bartholdi, E.; Ernst, R. R. (1976). "Two-dimensional spectroscopy. Application to nuclear magnetic resonance". Journal of Chemical Physics. 64 (5): 2229–46. Bibcode:1976JChPh..64.2229A. doi:10.1063/1.432450.
↑ Mateescu, Gheorghe D.; Valeriu, Adrian (1993). 2D NMR Density Matrix and Product Operator Treatment. Englewood Cliffs, New Jersey: PTR Prentice Hall.
↑ Akitt, J. W.; Mann, B. E. (2000). NMR and Chemistry. Cheltenham, UK: Stanley Thornes. p.273.
↑ Buddrus, J. and Bauer, H. (1987), Direct Identification of the Carbon Skeleton of Organic Compounds using Double Quantum Coherence 13C-NMR Spectroscopy. The INADEQUATE Pulse Sequence. Angew. Chem. Int. Ed. Engl., 26: 625-642. https://doi.org/10.1002/anie.198706251
1 2 Nakanishi, Koji, ed. (1990). One-dimensional and two-dimensional NMR Spectra by Modern Pulse Techniques. Mill Valley, California: University Science Books. p.136. ISBN0-935702-63-6.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.