Principles
Electronic transitions are typically observed in the visible and ultraviolet regions, in the wavelength range approximately 200–700 nm (50,000–14,000 cm−1), whereas fundamental vibrations are observed below about 4000 cm−1. [note 1] When the electronic and vibrational energy changes are so different, vibronic coupling (mixing of electronic and vibrational wave functions) can be neglected and the energy of a vibronic level can be taken as the sum of the electronic and vibrational (and rotational) energies; that is, the Born–Oppenheimer approximation applies. [4] The overall molecular energy depends not only on the electronic state but also on vibrational and rotational quantum numbers, denoted v and J respectively for diatomic molecules. It is conventional to add a double prime (v″, J″) for levels of the electronic ground state and a single prime (v′, J′) for electronically excited states.
Each electronic transition may show vibrational coarse structure, and for molecules in the gas phase, rotational fine structure. This is true even when the molecule has a zero dipole moment and therefore has no vibration-rotation infrared spectrum or pure rotational microwave spectrum. [5]
It is necessary to distinguish between absorption and emission spectra. With absorption the molecule starts in the ground electronic state, and usually also in the vibrational ground state v″ = 0 because at ordinary temperatures the energy necessary for vibrational excitation is large compared to the average thermal energy. The molecule is excited to another electronic state and to many possible vibrational states v' = 0, 1, 2, 3, .... With emission, the molecule can start in various populated vibrational states, and finishes in the electronic ground state in one of many populated vibrational levels. The emission spectrum is more complicated than the absorption spectrum of the same molecule because there are more changes in vibrational energy level.


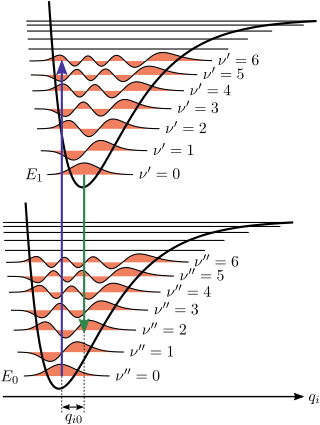
For absorption spectra, the vibrational coarse structure for a given electronic transition forms a single progression, or series of transitions with a common level, here the lower level v″ = 0. [6] There are no selection rules for vibrational quantum numbers, which are zero in the ground vibrational level of the initial electronic ground state, but can take any integer values in the final electronic excited state. The term values G(v) for a harmonic oscillator are given by where v is a vibrational quantum number, and ωe is the harmonic wavenumber. In the next approximation the term values are given by where χe is an anharmonicity constant. This is, in effect, a better approximation to the Morse potential near the potential minimum. The spacing between adjacent vibrational lines decreases with increasing quantum number because of anharmonicity in the vibration. Eventually the separation decreases to zero when the molecule photo-dissociates into a continuum of states. The second formula is adequate for small values of the vibrational quantum number. For higher values further anharmonicity terms are needed as the molecule approaches the dissociation limit, at the energy corresponding to the upper (final state) potential curve at infinite internuclear distance.


The intensity of allowed vibronic transitions is governed by the Franck–Condon principle. [7] The intensity distribution within a progression is governed by the difference in the equilibrium bond lengths of the initial electronic ground state and the final electronic excited state of the molecule. In accordance with the Born-Oppenheimer approximation, where electronic motion is near instantaneous compared to nuclear motion, transitions between vibrational levels happen with essentially no change in nuclear coordinates between the ground and excited electronic states. These nuclear coordinates are referred to as classical "turning points", where the equilibrium bond lengths of the initial and final electronic states are equal. [8] These transitions can be represented as vertical lines between the various vibrational levels within electronic states on an energy level diagram.
It is generally true that the greater the changes to the bond length of a molecule upon excitation, the greater the contribution of vibrational states to a progression. The width of this progression itself is dependent on the range of transition energies available for internuclear distances close to the turning points of the initial vibration state. As the "well" of the potential energy curve of the final electronic state grows steeper, there are more final vibrational states available for transitions, and thus more energy levels to yield a wider spectrum.
Emission spectra are complicated due to the variety of processes through which electronically excited molecules can spontaneously return to lower energy states. [9] There is a tendency for molecules to undergo vibrational energy relaxation, where energy is lost non-radiatively from the Franck–Condon state (the vibrational state achieved after a vertical transition) to surroundings or to internal processes. The molecules can settle in the ground vibrational level of the excited electronic state, where they can continue to decay to various vibrational levels in the ground electronic state, before ultimately returning to the lowest vibrational level of the ground state. [10]

If emission occurs before vibrational relaxation can occur, then the resulting fluorescence is referred to as resonance fluorescence. In this case, the emission spectrum is identical to the absorbance spectrum. Resonance fluorescence, however, is not very common and is mainly observed in small molecules (such as diatomics) in the gas phase. This lack of prevalence is due to short radiative lifetimes of the excited state, during which energy can be lost. [11] Emission from the ground vibrational level of the excited state after vibrational relaxation is much more prevalent, referred to as relaxed fluorescence. Emission peaks for a molecule exhibiting relaxed fluorescence are found at longer wavelengths than the corresponding absorption spectra, with the difference being the Stokes shift of the molecule.
Vibronic spectra of diatomic molecules in the gas phase have been analyzed in detail. [12] Vibrational coarse structure can sometimes be observed in the spectra of molecules in liquid or solid phases and of molecules in solution. Related phenomena including photoelectron spectroscopy, resonance Raman spectroscopy, luminescence, and fluorescence are not discussed in this article, though they also involve vibronic transitions.