
Androgen insensitivity syndrome (AIS) is a difference in sex development involving hormonal resistance due to androgen receptor dysfunction.

Leigh syndrome is an inherited neurometabolic disorder that affects the central nervous system. It is named after Archibald Denis Leigh, a British neuropsychiatrist who first described the condition in 1951. Normal levels of thiamine, thiamine monophosphate, and thiamine diphosphate are commonly found, but there is a reduced or absent level of thiamine triphosphate. This is thought to be caused by a blockage in the enzyme thiamine-diphosphate kinase, and therefore treatment in some patients would be to take thiamine triphosphate daily.

Lipoid congenital adrenal hyperplasia is an endocrine disorder that is an uncommon and potentially lethal form of congenital adrenal hyperplasia (CAH). It arises from defects in the earliest stages of steroid hormone synthesis: the transport of cholesterol into the mitochondria and the conversion of cholesterol to pregnenolone—the first step in the synthesis of all steroid hormones. Lipoid CAH causes mineralocorticoid deficiency in affected infants and children. Male infants are severely undervirilized causing their external genitalia to look feminine. The adrenals are large and filled with lipid globules derived from cholesterol.

Persistent Müllerian duct syndrome (PMDS) is the presence of Müllerian duct derivatives in what would be considered a genetically and otherwise physically normal male animal by typical human based standards. In humans, PMDS typically is due to an autosomal recessive congenital disorder and is considered by some to be a form of pseudohermaphroditism due to the presence of Müllerian derivatives.
Müllerian agenesis, also known as Müllerian aplasia, vaginal agenesis, or Mayer–Rokitansky–Küster–Hauser syndrome, is a congenital malformation characterized by a failure of the Müllerian ducts to develop, resulting in a missing uterus and variable degrees of vaginal hypoplasia of its upper portion. Müllerian agenesis is the cause in 15% of cases of primary amenorrhoea. Because most of the vagina does not develop from the Müllerian duct, instead developing from the urogenital sinus, along with the bladder and urethra, it is present even when the Müllerian duct is completely absent. Because ovaries do not develop from the Müllerian ducts, affected people might have normal secondary sexual characteristics but are infertile due to the lack of a functional uterus. However, biological motherhood is possible through uterus transplantation or use of gestational surrogates.

Coffin–Lowry syndrome is a genetic disorder that is X-linked dominant and which causes severe mental problems sometimes associated with abnormalities of growth, cardiac abnormalities, kyphoscoliosis, as well as auditory and visual abnormalities.
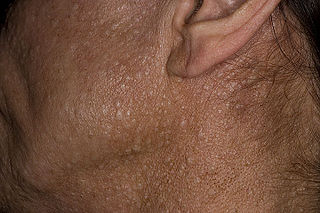
Birt–Hogg–Dubé syndrome (BHD), also Hornstein–Birt–Hogg–Dubé syndrome, Hornstein–Knickenberg syndrome, and fibrofolliculomas with trichodiscomas and acrochordons is a human, adult onset, autosomal dominant genetic disorder caused by the FLCN gene. It can cause susceptibility to kidney cancer, renal and pulmonary cysts, and noncancerous tumors of the hair follicles, called fibrofolliculomas. The symptoms seen in each family are unique, and can include any combination of the three symptoms. Fibrofolliculomas are the most common manifestation, found on the face and upper trunk in over 80% of people with BHD over the age of 40. Pulmonary cysts are equally common (84%) and 24% of people with BHD eventually experience a collapsed lung. Kidney tumors, both cancerous and benign, occur in 14–34% of people with BHD; the associated kidney cancers are often rare hybrid tumors.
Vaginal atresia is a condition in which the vagina is abnormally closed or absent. The main causes can either be complete vaginal hypoplasia, or a vaginal obstruction, often caused by an imperforate hymen or, less commonly, a transverse vaginal septum. It results in uterovaginal outflow tract obstruction. This condition does not usually occur by itself within an individual, but coupled with other developmental disorders within the female. The disorders that are usually coupled with a female who has vaginal atresia are Mayer-Rokitansky-Küster-Hauser syndrome, Bardet-Biedl syndrome, or Fraser syndrome. One out of every 5,000 women have this abnormality.

Larsen syndrome (LS) is a congenital disorder discovered in 1950 by Larsen and associates when they observed dislocation of the large joints and face anomalies in six of their patients. Patients with Larsen syndrome normally present with a variety of symptoms, including congenital anterior dislocation of the knees, dislocation of the hips and elbows, flattened facial appearance, prominent foreheads, and depressed nasal bridges. Larsen syndrome can also cause a variety of cardiovascular and orthopedic abnormalities. This rare disorder is caused by a genetic defect in the gene encoding filamin B, a cytoplasmic protein that is important in regulating the structure and activity of the cytoskeleton. The gene that influences the emergence of Larsen syndrome is found in chromosome region, 3p21.1-14.1, a region containing human type VII collagen gene. Larsen syndrome has recently been described as a mesenchyme disorder that affects the connective tissue of an individual. Autosomal dominant and recessive forms of the disorder have been reported, although most cases are autosomal dominant. Reports have found that in Western societies, Larsen syndrome can be found in one in every 100,000 births, but this is most likely an underestimate because the disorder is frequently unrecognized or misdiagnosed.

Alström syndrome (AS), also called Alström–Hallgren syndrome, is a very rare autosomal recessive genetic disorder characterised by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss and dilated cardiomyopathy. Endocrine disorders typically also occur, such as hypergonadotrophic hypogonadism and hypothyroidism, as well as acanthosis nigricans resulting from hyperinsulinemia. Developmental delay is seen in almost half of people with Alström syndrome.
XX gonadal dysgenesis is a type of female hypogonadism in which the ovaries do not function to induce puberty in an otherwise normal girl whose karyotype is found to be 46,XX. With nonfunctional streak ovaries, she is low in estrogen levels (hypoestrogenic) and has high levels of FSH and LH. Estrogen and progesterone therapy is usually then commenced. Some cases are considered a severe version of premature ovarian failure where the ovaries fail before puberty.

Disorders of sex development (DSDs), also known as differences in sex development, diverse sex development and variations in sex characteristics (VSC), are congenital conditions affecting the reproductive system, in which development of chromosomal, gonadal, or anatomical sex is atypical.

WNT4 is a secreted protein that, in humans, is encoded by the WNT4 gene, found on chromosome 1. It promotes female sex development and represses male sex development. Loss of function may have consequences, such as female to male sex reversal.
Complete androgen insensitivity syndrome (CAIS) is an AIS condition that results in the complete inability of the cell to respond to androgens. As such, the insensitivity to androgens is only clinically significant when it occurs in individuals who are exposed to significant amounts of testosterone at some point in their lives. The unresponsiveness of the cell to the presence of androgenic hormones prevents the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty, but does allow, without significant impairment, female genital and sexual development in those with the condition.
Leydig cell hypoplasia (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 genetic males. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism, hypergonadotropic hypogonadism, reduced or absent puberty, and infertility.
Müllerian duct anomalies are those structural anomalies caused by errors in Müllerian duct development during embryonic morphogenesis. Factors that precipitate include genetics, and maternal exposure to teratogens.
Acromesomelic dysplasia is a rare skeletal disorder that causes abnormal bone and cartilage development, leading to shortening of the forearms, lower legs, hands, feet, fingers, and toes. Five different genetic mutations have been implicated in the disorder. Treatment is individualized but is generally aimed at palliating symptoms, for example, treatment of kyphosis and lumbar hyperlordosis.

Vaginal anomalies are abnormal structures that are formed during the prenatal development of the female reproductive system and are rare congenital defects that result in an abnormal or absent vagina.

SRD5A3-CDG is a rare, non X-linked congenital disorder of glycosylation (CDG) due to a mutation in the steroid 5 alpha reductase type 3 gene. It is one of over 150 documented types of Congenital disorders of Glycosylation. Like many other CDGs, SRD5A3 is ultra-rare, with around 38 documented cases in the world.

17q12 microdeletion syndrome, also known as 17q12 deletion syndrome, is a rare chromosomal anomaly caused by the deletion of a small amount of material from a region in the long arm of chromosome 17. It is typified by deletion of the HNF1B gene, resulting in kidney abnormalities and renal cysts and diabetes syndrome. It also has neurocognitive effects, and has been implicated as a genetic factor for autism and schizophrenia.

