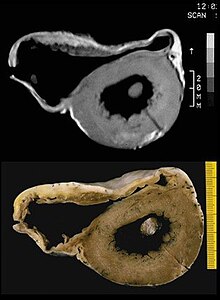
Cardiomyopathy is a group of primary diseases of the heart muscle. Early on there may be few or no symptoms. As the disease worsens, shortness of breath, feeling tired, and swelling of the legs may occur, due to the onset of heart failure. An irregular heart beat and fainting may occur. Those affected are at an increased risk of sudden cardiac death.

A premature ventricular contraction (PVC) is a common event where the heartbeat is initiated by Purkinje fibers in the ventricles rather than by the sinoatrial node. PVCs may cause no symptoms or may be perceived as a "skipped beat" or felt as palpitations in the chest. PVCs do not usually pose any danger.

Palpitations are perceived abnormalities of the heartbeat characterized by awareness of cardiac muscle contractions in the chest, which is further characterized by the hard, fast and/or irregular beatings of the heart.

A desmosome, also known as a macula adherens, is a cell structure specialized for cell-to-cell adhesion. A type of junctional complex, they are localized spot-like adhesions randomly arranged on the lateral sides of plasma membranes. Desmosomes are one of the stronger cell-to-cell adhesion types and are found in tissue that experience intense mechanical stress, such as cardiac muscle tissue, bladder tissue, gastrointestinal mucosa, and epithelia.

Dilated cardiomyopathy (DCM) is a condition in which the heart becomes enlarged and cannot pump blood effectively. Symptoms vary from none to feeling tired, leg swelling, and shortness of breath. It may also result in chest pain or fainting. Complications can include heart failure, heart valve disease, or an irregular heartbeat.

Ventricular tachycardia is a cardiovascular disorder in which fast heart rate occurs in the ventricles of the heart. Although a few seconds of VT may not result in permanent problems, longer periods are dangerous; and multiple episodes over a short period of time are referred to as an electrical storm. Short periods may occur without symptoms, or present with lightheadedness, palpitations, shortness of breath, chest pain, and decreased level of consciousness. Ventricular tachycardia may lead to coma and persistent vegetative state due to lack of blood and oxygen to the brain. Ventricular tachycardia may result in ventricular fibrillation (VF) and turn into cardiac arrest. This conversion of the VT into VF is called the degeneration of the VT. It is found initially in about 7% of people in cardiac arrest.

Desmin is a protein that in humans is encoded by the DES gene. Desmin is a muscle-specific, type III intermediate filament that integrates the sarcolemma, Z disk, and nuclear membrane in sarcomeres and regulates sarcomere architecture.

Restrictive cardiomyopathy (RCM) is a form of cardiomyopathy in which the walls of the heart are rigid. Thus the heart is restricted from stretching and filling with blood properly. It is the least common of the three original subtypes of cardiomyopathy: hypertrophic, dilated, and restrictive.
Desmoglein-2 is a protein that in humans is encoded by the DSG2 gene. Desmoglein-2 is highly expressed in epithelial cells and cardiomyocytes. Desmoglein-2 is localized to desmosome structures at regions of cell-cell contact and functions to structurally adhere adjacent cells together. In cardiac muscle, these regions are specialized regions known as intercalated discs. Mutations in desmoglein-2 have been associated with arrhythmogenic right ventricular cardiomyopathy and familial dilated cardiomyopathy.

Desmoplakin is a protein in humans that is encoded by the DSP gene. Desmoplakin is a critical component of desmosome structures in cardiac muscle and epidermal cells, which function to maintain the structural integrity at adjacent cell contacts. In cardiac muscle, desmoplakin is localized to intercalated discs which mechanically couple cardiac cells to function in a coordinated syncytial structure. Mutations in desmoplakin have been shown to play a role in dilated cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy, where it may present with acute myocardial injury; striate palmoplantar keratoderma, Carvajal syndrome and paraneoplastic pemphigus.

Plakoglobin, also known as junction plakoglobin or gamma-catenin, is a protein that in humans is encoded by the JUP gene. Plakoglobin is a member of the catenin protein family and homologous to β-catenin. Plakoglobin is a cytoplasmic component of desmosomes and adherens junctions structures located within intercalated discs of cardiac muscle that function to anchor sarcomeres and join adjacent cells in cardiac muscle. Mutations in plakoglobin are associated with arrhythmogenic right ventricular dysplasia.
Athletic heart syndrome (AHS) is a non-pathological condition commonly seen in sports medicine in which the human heart is enlarged, and the resting heart rate is lower than normal.

Noncompaction cardiomyopathy (NCC) is a rare congenital disease of heart muscle that affects both children and adults. It results from abnormal prenatal development of heart muscle.

Ryanodine receptor 2 (RYR2) is one of a class of ryanodine receptors and a protein found primarily in cardiac muscle. In humans, it is encoded by the RYR2 gene. In the process of cardiac calcium-induced calcium release, RYR2 is the major mediator for sarcoplasmic release of stored calcium ions.

Desmocollin-2 is a protein that in humans is encoded by the DSC2 gene. Desmocollin-2 is a cadherin-type protein that functions to link adjacent cells together in specialized regions known as desmosomes. Desmocollin-2 is widely expressed, and is the only desmocollin isoform expressed in cardiac muscle, where it localizes to intercalated discs. Mutations in DSC2 have been causally linked to arrhythmogenic right ventricular cardiomyopathy.

Plakophilin-2 is a protein that in humans is encoded by the PKP2 gene. Plakophilin 2 is expressed in skin and cardiac muscle, where it functions to link cadherins to intermediate filaments in the cytoskeleton. In cardiac muscle, plakophilin-2 is found in desmosome structures located within intercalated discs. Mutations in PKP2 have been shown to be causal in arrhythmogenic right ventricular cardiomyopathy.
Desmocollins are a subfamily of desmosomal cadherins, the transmembrane constituents of desmosomes. They are co-expressed with desmogleins to link adjacent cells by extracellular adhesion. There are seven desmosomal cadherins in humans, three desmocollins and four desmogleins. Desmosomal cadherins allow desmosomes to contribute to the integrity of tissue structure in multicellular living organisms.
Boxer cardiomyopathy is a disease of the myocardium primarily affecting Boxer dogs. It is characterized by the development of ventricular tachyarrhythmias, resulting in syncope and sudden cardiac death. Myocardial failure and congestive heart failure are uncommon manifestations of the disease.

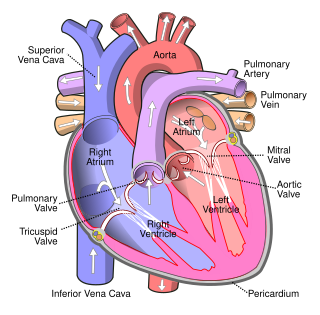
Arrhythmias, also known as cardiac arrhythmias, are irregularities in the heartbeat, including when it is too fast or too slow. A resting heart rate that is too fast – above 100 beats per minute in adults – is called tachycardia, and a resting heart rate that is too slow – below 60 beats per minute – is called bradycardia. Some types of arrhythmias have no symptoms. Symptoms, when present, may include palpitations or feeling a pause between heartbeats. In more serious cases, there may be lightheadedness, passing out, shortness of breath, chest pain, or decreased level of consciousness. While most cases of arrhythmia are not serious, some predispose a person to complications such as stroke or heart failure. Others may result in sudden death.
Frank I. Marcus was an American cardiologist and Emeritus Professor of Medicine at the University of Arizona Health Sciences Center, the author of more than 290 publications in peer-reviewed medical journals and of 90 book chapters. He was considered a world expert on arrhythmogenic right ventricular cardiomyopathy (ARVC) and was a member of the Editorial/Scientific Board of 14 Cardiovascular Journals as well as a reviewer for 26 other medical publications.