
Beta globulins are a group of globular proteins in plasma that are more mobile in alkaline or electrically charged solutions than gamma globulins, but less mobile than alpha globulins. [1] [2] [3]
Examples of beta globulins include:
Beta globulins are a group of globular proteins in plasma that are more mobile in alkaline or electrically charged solutions than gamma globulins, but less mobile than alpha globulins. [1] [2] [3]
Examples of beta globulins include:
The Structural Classification of Proteins (SCOP) database is a largely manual classification of protein structural domains based on similarities of their structures and amino acid sequences. A motivation for this classification is to determine the evolutionary relationship between proteins. Proteins with the same shapes but having little sequence or functional similarity are placed in different superfamilies, and are assumed to have only a very distant common ancestor. Proteins having the same shape and some similarity of sequence and/or function are placed in "families", and are assumed to have a closer common ancestor.
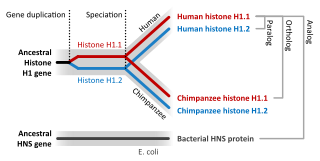
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).
In academia, computational immunology is a field of science that encompasses high-throughput genomic and bioinformatics approaches to immunology. The field's main aim is to convert immunological data into computational problems, solve these problems using mathematical and computational approaches and then convert these results into immunologically meaningful interpretations.
The DrugBank database is a comprehensive, freely accessible, online database containing information on drugs and drug targets created and maintained by the University of Alberta and The Metabolomics Innovation Centre located in Alberta, Canada. As both a bioinformatics and a cheminformatics resource, DrugBank combines detailed drug data with comprehensive drug target information. DrugBank has used content from Wikipedia; Wikipedia also often links to Drugbank, posing potential circular reporting issues.

Amos Bairoch is a Swiss bioinformatician and Professor of Bioinformatics at the Department of Human Protein Sciences of the University of Geneva where he leads the CALIPHO group at the Swiss Institute of Bioinformatics (SIB) combining bioinformatics, curation, and experimental efforts to functionally characterize human proteins.
Chemical Entities of Biological Interest, also known as ChEBI, is a chemical database and ontology of molecular entities focused on 'small' chemical compounds, that is part of the Open Biomedical Ontologies (OBO) effort at the European Bioinformatics Institute (EBI). The term "molecular entity" refers to any "constitutionally or isotopically distinct atom, molecule, ion, ion pair, radical, radical ion, complex, conformer, etc., identifiable as a separately distinguishable entity". The molecular entities in question are either products of nature or synthetic products which have potential bioactivity. Molecules directly encoded by the genome, such as nucleic acids, proteins and peptides derived from proteins by proteolytic cleavage, are not as a rule included in ChEBI.

PROSITE is a protein database. It consists of entries describing the protein families, domains and functional sites as well as amino acid patterns and profiles in them. These are manually curated by a team of the Swiss Institute of Bioinformatics and tightly integrated into Swiss-Prot protein annotation. PROSITE was created in 1988 by Amos Bairoch, who directed the group for more than 20 years. Since July 2018, the director of PROSITE and Swiss-Prot is Alan Bridge.

David J. Lipman is an American biologist who from 1989 to 2017 was the director of the National Center for Biotechnology Information (NCBI) at the National Institutes of Health. NCBI is the home of GenBank, the U.S. node of the International Sequence Database Consortium, and PubMed, one of the most heavily used sites in the world for the search and retrieval of biomedical information. Lipman is one of the original authors of the BLAST sequence alignment program, and a respected figure in bioinformatics. In 2017, he left NCBI and became Chief Science Officer at Impossible Foods.

MicrobesOnline is a publicly and freely accessible website that hosts multiple comparative genomic tools for comparing microbial species at the genomic, transcriptomic and functional levels. MicrobesOnline was developed by the Virtual Institute for Microbial Stress and Survival, which is based at the Lawrence Berkeley National Laboratory in Berkeley, California. The site was launched in 2005, with regular updates until 2011.

6C RNA is a class of non-coding RNA present in actinomycetes. 6C RNA was originally discovered as a conserved RNA structure having two stem-loops each containing six or more cytosine (C) residues. Later work revealed that 6C RNAs in Streptomyces coelicolor and Streptomyces avermitilis have predicted rho-independent transcription terminators, and microarray and reverse-transcriptase PCR experiments indicate that the S. coelicolor version is transcribed as RNA. Transcription of the S. coelicolor RNA increases during sporulation, and three transcripts were detected that overlap the 6C motif, but have different apparent start and stop sites.
This microRNA database and microRNA targets databases is a compilation of databases and web portals and servers used for microRNAs and their targets. MicroRNAs (miRNAs) represent an important class of small non-coding RNAs (ncRNAs) that regulate gene expression by targeting messenger RNAs.
Gypsy (GyDB) is a wiki-style database of mobile genetic elements.
The eggNOG database is a database of biological information hosted by the EMBL. It is based on the original idea of COGs and expands that idea to non-supervised orthologous groups constructed from numerous organisms. The database was created in 2007 and updated to version 4.5 in 2015. eggNOG stands for evolutionary genealogy of genes: Non-supervised Orthologous Groups.
The Human Metabolome Database (HMDB) is a comprehensive, high-quality, freely accessible, online database of small molecule metabolites found in the human body. Created by the Human Metabolome Project funded by Genome Canada. One of the first dedicated metabolomics databases, the HMDB facilitates human metabolomics research, including the identification and characterization of human metabolites using NMR spectroscopy, GC-MS spectrometry and LC/MS spectrometry. To aid in this discovery process, the HMDB contains three kinds of data: 1) chemical data, 2) clinical data, and 3) molecular biology/biochemistry data. The chemical data includes 41,514 metabolite structures with detailed descriptions along with nearly 10,000 NMR, GC-MS and LC/MS spectra.
MetaboAnalyst is a set of online tools for metabolomic data analysis and interpretation, created by members of the Wishart Research Group at the University of Alberta. It was first released in May 2009 and version 2.0 was released in January 2012. MetaboAnalyst provides a variety of analysis methods that have been tailored for metabolomic data. These methods include metabolomic data processing, normalization, multivariate statistical analysis, and data annotation. The current version is focused on biomarker discovery and classification.
The Histone Database is a comprehensive database of histone protein sequences including histone variants, classified by histone types and variants, maintained by National Center for Biotechnology Information. The creation of the Histone Database was stimulated by the X-ray analysis of the structure of the nucleosomal core histone octamer followed by the application of a novel motif searching method to a group of proteins containing the histone fold motif in the early-mid-1990. The first version of the Histone Database was released in 1995 and several updates have been released since then.
Non-coding RNAs have been discovered using both experimental and bioinformatic approaches. Bioinformatic approaches can be divided into three main categories. The first involves homology search, although these techniques are by definition unable to find new classes of ncRNAs. The second category includes algorithms designed to discover specific types of ncRNAs that have similar properties. Finally, some discovery methods are based on very general properties of RNA, and are thus able to discover entirely new kinds of ncRNAs.

An array of protein tandem repeats is defined as several adjacent copies having the same or similar sequence motifs. These periodic sequences are generated by internal duplications in both coding and non-coding genomic sequences. Repetitive units of protein tandem repeats are considerably diverse, ranging from the repetition of a single amino acid to domains of 100 or more residues.
| | This cell biology article is a stub. You can help Wikipedia by expanding it. |