
A sarcoma is a malignant tumor, a type of cancer that arises from cells of mesenchymal origin. Connective tissue is a broad term that includes bone, cartilage, muscle, fat, vascular, or other structural tissues, and sarcomas can arise in any of these types of tissues. As a result, there are many subtypes of sarcoma, which are classified based on the specific tissue and type of cell from which the tumor originates.

Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal neoplasms of the gastrointestinal tract. GISTs arise in the smooth muscle pacemaker interstitial cell of Cajal, or similar cells. They are defined as tumors whose behavior is driven by mutations in the KIT gene (85%), PDGFRA gene (10%), or BRAF kinase (rare). 95% of GISTs stain positively for KIT (CD117). Most (66%) occur in the stomach and gastric GISTs have a lower malignant potential than tumors found elsewhere in the GI tract.

Dermatofibrosarcoma protuberans (DFSP) is a rare locally aggressive malignant cutaneous soft-tissue sarcoma. DFSP develops in the connective tissue cells in the middle layer of the skin (dermis). Estimates of the overall occurrence of DFSP in the United States are 0.8 to 4.5 cases per million persons per year. In the United States, DFSP accounts for between 1 and 6 percent of all soft-tissue sarcomas and 18 percent of all cutaneous soft-tissue sarcomas. In the Surveillance, Epidemiology and End Results (SEER) tumor registry from 1992 through 2004, DFSP was second only to Kaposi sarcoma.

Liposarcomas are the most common subtype of soft tissue sarcomas, accounting for at least 20% of all sarcomas in adults. Soft tissue sarcomas are rare neoplasms with over 150 different histological subtypes or forms. Liposarcomas arise from the precursor lipoblasts of the adipocytes in adipose tissues. Adipose tissues are distributed throughout the body, including such sites as the deep and more superficial layers of subcutaneous tissues as well as in less surgically accessible sites like the retroperitoneum and visceral fat inside the abdominal cavity.

Desmoplastic small-round-cell tumor (DSRCT) is an aggressive and rare cancer that primarily occurs as masses in the abdomen. Other areas affected may include the lymph nodes, the lining of the abdomen, diaphragm, spleen, liver, chest wall, skull, spinal cord, large intestine, small intestine, bladder, brain, lungs, testicles, ovaries, and the pelvis. Reported sites of metastatic spread include the liver, lungs, lymph nodes, brain, skull, and bones. It is characterized by the EWS-WT1 fusion protein.
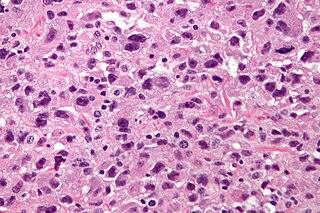
Undifferentiated pleomorphic sarcoma (UPS), also termed pleomorphic myofibrosarcoma, high-grade myofibroblastic sarcoma, and high-grade myofibrosarcoma, is characterized by the World Health Organization (WHO) as a rare, poorly differentiated neoplasm. WHO classified it as one of the undifferentiated/unclassified sarcomas in the category of tumors of uncertain differentiation. Sarcomas are cancers derived mesenchymal stem cells that typically develop in bone, muscle, fat, blood vessels, lymphatic vessels, tendons, and ligaments. More than 70 sarcoma subtypes have been described. The UPS subtype of these sarcomas consists of tumor cells that are poorly differentiated and may appear as spindle-shaped cells, histiocytes, and giant cells. UPS is considered a diagnosis that defies formal sub-classification after thorough histologic, immunohistochemical, and ultrastructural examinations fail to identify the type of cells involved.

Ewing sarcoma is a type of pediatric cancer that forms in bone or soft tissue. Symptoms may include swelling and pain at the site of the tumor, fever, and a bone fracture. The most common areas where it begins are the legs, pelvis, and chest wall. In about 25% of cases, the cancer has already spread to other parts of the body at the time of diagnosis. Complications may include a pleural effusion or paraplegia.

Congenital mesoblastic nephroma, while rare, is the most common kidney neoplasm diagnosed in the first three months of life and accounts for 3-5% of all childhood renal neoplasms. This neoplasm is generally non-aggressive and amenable to surgical removal. However, a readily identifiable subset of these kidney tumors has a more malignant potential and is capable of causing life-threatening metastases. Congenital mesoblastic nephroma was first named as such in 1967 but was recognized decades before this as fetal renal hamartoma or leiomyomatous renal hamartoma.
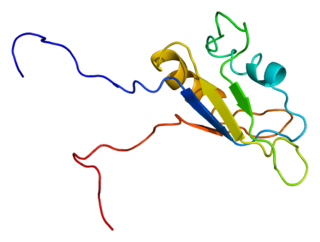
RNA-binding protein EWS is a protein that in humans is encoded by the EWSR1 gene on human chromosome 22, specifically 22q12.2. It is one of 3 proteins in the FET protein family.

Epithelioid sarcoma is a rare soft tissue sarcoma arising from mesenchymal tissue and characterized by epithelioid-like features. It accounts for less than 1% of all soft tissue sarcomas. It was first definitively characterized by F.M. Enzinger in 1970. It commonly presents itself in the distal limbs of young adults as a small, soft mass or a cluster of bumps. A proximal version has also been described, frequently occurring in the upper extremities. Less commonly, cases are reported in the pelvis, vulva, penis, and spine.
Extraskeletal myxoid chondrosarcoma (EMC) is a rare low-grade malignant mesenchymal neoplasm of the soft tissues, that differs from other sarcomas by unique histology and characteristic chromosomal translocations. There is an uncertain differentiation and neuroendocrine differentiation is even possible.
Porocarcinoma (PCA) is a rare form of skin cancer that develops in eccrine sweat glands, i.e. the body's widely distributed major type of sweat glands, as opposed to the apocrine sweat glands which are located primarily in the armpits and perineal area. This cancer typically develops in individuals as a single cutaneous tumor in the intraepidermal spiral part of these sweat glands' ducts at or near to where they open on the skin's surface. PCA tumors are classified as one form of the cutaneous adnexal tumors; in a study of 2,205 cases, PCA was the most common (11.8%) form of these tumors.

Spiradenomas (SA) are rare, benign cutaneous adnexal tumors that may progress to become their malignant counterparts, i.e. spiradenocarcinomas (SAC). Cutaneous adnexal tumors are a group of skin tumors consisting of tissues that have differentiated towards one of the four primary adnexal structures found in normal skin: hair follicles, sebaceous sweat glands, apocrine sweat glands, and eccrine sweat glands. SA and SAC tumors were regarded as eccrine gland tumors and termed eccrine spiradenomas and eccrine spiradenocarcinomas, respectively. However, more recent studies have found them to be hair follicle tumors and commonly term them spiradenomas and spiradenocarcinomas, respectively. Further confusing the situation, SA-like and SAC-like tumors are also 1) manifestations of the inherited disorder, CYLD cutaneous syndrome (CCS), and 2) have repeatedly been confused with an entirely different tumor, adenoid cystic carcinomas of the salivary gland. Here, SA and SAC are strictly defined as sporadic hair follicle tumors that do not include the hereditary CCS spiradenomas and heridtary spiradenocarcinoms of CCS or the adenoid cystic carcinomas.

Low-grade fibromyxoid sarcoma (LGFMS) is a rare type of low-grade sarcoma first described by H. L. Evans in 1987. LGFMS are soft tissue tumors of the mesenchyme-derived connective tissues; on microscopic examination, they are found to be composed of spindle-shaped cells that resemble fibroblasts. These fibroblastic, spindle-shaped cells are neoplastic cells that in most cases of LGFMS express fusion genes, i.e. genes composed of parts of two different genes that form as a result of mutations. The World Health Organization (2020) classified LGFMS as a specific type of tumor in the category of malignant fibroblastic and myofibroblastic tumors.
Acral myxoinflammatory fibroblastic sarcoma (AMSF), also termed myxoinflammatory fibroblastic sarcoma (MSF), is a rare, low-grade, soft tissue tumor that the World Health Organization (2020) classified as in the category of rarely metastasizing fibroblastic and myofibroblastic tumors. It is a locally aggressive neoplasm that often recurs at the site of its surgical removal. However, it usually grows slowly and in only 1–2% of cases spreads to distant tissues.
Fibroblastic and myofibroblastic tumors (FMTs) are tumors which develop from the mesenchymal stem cells which differentiate into fibroblasts and/or the myocytes/myoblasts that differentiate into muscle cells. FMTs are a heterogeneous group of soft tissue neoplasms. The World Health Organization (2020) defined tumors as being FMTs based on their morphology and, more importantly, newly discovered abnormalities in the expression levels of key gene products made by these tumors' neoplastic cells. Histopathologically, FMTs consist of neoplastic connective tissue cells which have differented into cells that have microscopic appearances resembling fibroblasts and/or myofibroblasts. The fibroblastic cells are characterized as spindle-shaped cells with inconspicuous nucleoli that express vimentin, an intracellular protein typically found in mesenchymal cells, and CD34, a cell surface membrane glycoprotein. Myofibroblastic cells are plumper with more abundant cytoplasm and more prominent nucleoli; they express smooth muscle marker proteins such as smooth muscle actins, desmin, and caldesmon. The World Health Organization further classified FMTs into four tumor forms based on their varying levels of aggressiveness: benign, intermediate, intermediate, and malignant.
Myxofibrosarcoma (MFS), although a rare type of tumor, is one of the most common soft tissue sarcomas, i.e. cancerous tumors, that develop in the soft tissues of elderly individuals. Initially considered to be a type of histiocytoma termed fibrous histiocytoma or myxoid variant of malignant fibrous histiocytoma, Angervall et al. termed this tumor myxofibrosarcoma in 1977. In 2020, the World Health Organization reclassified MFS as a separate and distinct tumor in the category of malignant fibroblastic and myofibroblastic tumors.
Sclerosing epithelioid fibrosarcoma (SEF) is a very rare malignant tumor of soft tissues that on microscopic examination consists of small round or ovoid neoplastic epithelioid fibroblast-like cells, i.e. cells that have features resembling both epithelioid cells and fibroblasts. In 2020, the World Health Organization classified SEF as a distinct tumor type in the category of malignant fibroblastic and myofibroblastic tumors. However, current studies have reported that low-grade fibromyxoid sarcoma (LGFMS) has many clinically and pathologically important features characteristic of SEF; these studies suggest that LGSFMS may be an early form of, and over time progress to become, a SEF. Since the World Health Organization has classified LGFMS as one of the malignant fibroblastic and myofibroblastic tumors that is distinctly different than SEF, SEF and LGFMS are here regarded as different tumor forms.
The FET protein family consists of three similarly structured and functioning proteins. They and the genes in the FET gene family which encode them are: 1) the EWSR1 protein encoded by the EWSR1 gene located at band 12.2 of the long arm of chromosome 22; 2) the FUS protein encoded by the FUS gene located at band 16 on the short arm of chromosome 16; and 3) the TAF15 protein encoded by the TAF15 gene located at band 12 on the long arm of chromosome 7 The FET in this protein family's name derives from the first letters of FUS, EWSR1, and TAF15.
Low-grade myofibroblastic sarcoma (LGMS) is a subtype of the malignant sarcomas. As it is currently recognized, LGMS was first described as a rare, atypical myofibroblastic tumor by Mentzel et al. in 1998. Myofibroblastic sarcomas had been divided into low-grade myofibroblastic sarcomas, intermediate‐grade myofibroblasic sarcomas, i.e. IGMS, and high‐grade myofibroblasic sarcomas, i.e. HGMS based on their microscopic morphological, immunophenotypic, and malignancy features. LGMS and IGMS are now classified together by the World Health Organization (WHO), 2020, in the category of intermediate fibroblastic and myofibroblastic tumors. WHO, 2020, classifies HGMS as a soft tissue tumor in the category of tumors of uncertain differentiation. This article follows the WHO classification: here, LGMS includes IGMS but not HGMS which is a more aggressive and metastasizing tumor than LGMS and consists of cells of uncertain origin.
