Deamidation is a chemical reaction in which an amide functional group in the side chain of the amino acids asparagine or glutamine is removed or converted to another functional group. Typically, asparagine is converted to aspartic acid or isoaspartic acid. Glutamine is converted to glutamic acid or pyroglutamic acid (5-oxoproline). In a protein or peptide, these reactions are important because they may alter its structure, stability or function and may lead to protein degradation. The net chemical change is the addition of a water group and removal of an ammonia group, which corresponds to a +1 (0.98402) Da mass increase. Although deamidation occurs on glutamine, glycosylated asparagine and other amides, these are negligible under typical proteolysis conditions. [1]
Contents
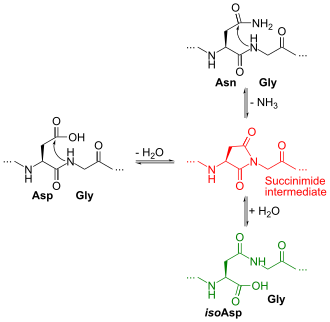
In the deamidation of an asparagine residue under physiological conditions, the side chain is attacked by the nitrogen atom of the following peptide group (in black at top right of Figure), forming an asymmetric succinimide intermediate (in red). The asymmetry of the intermediate results in two products of its hydrolysis, either aspartic acid (in black at left) or isoaspartic acid, which is a beta amino acid (in green at bottom right). However, there is a concern that aspartic acid can be isomerized after deamidation. [2] The deamidation of a glutamine residue may proceed via the same mechanism but at a much slower rate since formation of the six-member-ring glutarimide intermediate is less favoured than the succinimide intermediate for asparagine. In general, deamidation can be eliminated by proteolysis at an acidic pH or at a slightly basic pH (4.5 and 8.0, respectively) using the endoprotease, Glu-C. [2]
The rates of deamidation depend on multiple factors, including the primary sequences and higher-order structures of the proteins, pH, temperature, and components in the solutions. Most potential deamidation sites are stabilized by higher order structure. Asn-Gly (NG), is the most flexible and since it is acidic, it is most prone to deamidation with a half-life around 24 h under physiological conditions (pH 7.4, 37 °C). [3]
As a free amino acid, or as the N-terminal residue of a peptide or protein, glutamine deamidates readily to form pyroglutamic acid (5-oxoproline). The reaction proceeds via nucleophilic attack of the α-amino group on the side-chain amide to form a γ-lactam with the elimination of ammonia from the side-chain.