
The brachiocephalic artery, brachiocephalic trunk, or innominate artery is an artery of the mediastinum that supplies blood to the right arm, head, and neck.

Patent ductus arteriosus (PDA) is a medical condition in which the ductus arteriosus fails to close after birth: this allows a portion of oxygenated blood from the left heart to flow back to the lungs through the aorta, which has a higher blood pressure, to the pulmonary artery, which has a lower blood pressure. Symptoms are uncommon at birth and shortly thereafter, but later in the first year of life there is often the onset of an increased work of breathing and failure to gain weight at a normal rate. With time, an uncorrected PDA usually leads to pulmonary hypertension followed by right-sided heart failure.
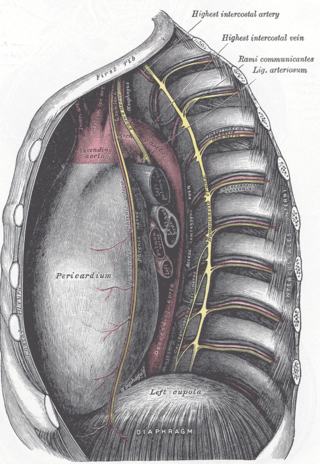
The ligamentum arteriosum, also known as Botallo's ligament, Harvey's ligament, and Botallo's duct, is a small ligament attaching the aorta to the pulmonary artery. It serves no function in adults but is the remnant of the ductus arteriosus formed within three weeks after birth.

dextro-Transposition of the great arteries is a potentially life-threatening birth defect in the large arteries of the heart. The primary arteries are transposed.

The ductus arteriosus, also called the ductus Botalli, named after the Italian physiologist Leonardo Botallo, is a blood vessel in the developing fetus connecting the trunk of the pulmonary artery to the proximal descending aorta. It allows most of the blood from the right ventricle to bypass the fetus's fluid-filled non-functioning lungs. Upon closure at birth, it becomes the ligamentum arteriosum.

Coarctation of the aorta (CoA) is a congenital condition whereby the aorta is narrow, usually in the area where the ductus arteriosus inserts. The word coarctation means "pressing or drawing together; narrowing". Coarctations are most common in the aortic arch. The arch may be small in babies with coarctations. Other heart defects may also occur when coarctation is present, typically occurring on the left side of the heart. When a patient has a coarctation, the left ventricle has to work harder. Since the aorta is narrowed, the left ventricle must generate a much higher pressure than normal in order to force enough blood through the aorta to deliver blood to the lower part of the body. If the narrowing is severe enough, the left ventricle may not be strong enough to push blood through the coarctation, thus resulting in a lack of blood to the lower half of the body. Physiologically its complete form is manifested as interrupted aortic arch.

The recurrent laryngeal nerve (RLN) is a branch of the vagus nerve that supplies all the intrinsic muscles of the larynx, with the exception of the cricothyroid muscles. There are two recurrent laryngeal nerves, right and left. The right and left nerves are not symmetrical, with the left nerve looping under the aortic arch, and the right nerve looping under the right subclavian artery, then traveling upwards. They both travel alongside the trachea. Additionally, the nerves are among the few nerves that follow a recurrent course, moving in the opposite direction to the nerve they branch from, a fact from which they gain their name.

Persistent truncus arteriosus (PTA), often referred to simply as truncus arteriosus, is a rare form of congenital heart disease that presents at birth. In this condition, the embryological structure known as the truncus arteriosus fails to properly divide into the pulmonary trunk and aorta. This results in one arterial trunk arising from the heart and providing mixed blood to the coronary arteries, pulmonary arteries, and systemic circulation. For the International Classification of Diseases (ICD-11), the International Paediatric and Congenital Cardiac Code (IPCCC) was developed to standardize the nomenclature of congenital heart disease. Under this system, English is now the official language, and persistent truncus arteriosus should properly be termed common arterial trunk.
The aortic arch, arch of the aorta, or transverse aortic arch is the part of the aorta between the ascending and descending aorta. The arch travels backward, so that it ultimately runs to the left of the trachea.

The aortic arches or pharyngeal arch arteries are a series of six paired embryological vascular structures which give rise to the great arteries of the neck and head. They are ventral to the dorsal aorta and arise from the aortic sac.

Aberrant subclavian artery, or aberrant subclavian artery syndrome, is a rare anatomical variant of the origin of the right or left subclavian artery. This abnormality is the most common congenital vascular anomaly of the aortic arch, occurring in approximately 1% of individuals.

Ortner's syndrome is a rare cardiovocal syndrome and refers to recurrent laryngeal nerve palsy from cardiovascular disease. It was first described by Norbert Ortner (1865–1935), an Austrian physician, in 1897.

Larsen syndrome (LS) is a congenital disorder discovered in 1950 by Larsen and associates when they observed dislocation of the large joints and face anomalies in six of their patients. Patients with Larsen syndrome normally present with a variety of symptoms, including congenital anterior dislocation of the knees, dislocation of the hips and elbows, flattened facial appearance, prominent foreheads, and depressed nasal bridges. Larsen syndrome can also cause a variety of cardiovascular and orthopedic abnormalities. This rare disorder is caused by a genetic defect in the gene encoding filamin B, a cytoplasmic protein that is important in regulating the structure and activity of the cytoskeleton. The gene that influences the emergence of Larsen syndrome is found in chromosome region, 3p21.1-14.1, a region containing human type VII collagen gene. Larsen syndrome has recently been described as a mesenchyme disorder that affects the connective tissue of an individual. Autosomal dominant and recessive forms of the disorder have been reported, although most cases are autosomal dominant. Reports have found that in Western societies, Larsen syndrome can be found in one in every 100,000 births, but this is most likely an underestimate because the disorder is frequently unrecognized or misdiagnosed.
Interrupted aortic arch is a very rare heart defect in which the aorta is not completely developed. There is a gap between the ascending and descending thoracic aorta. In a sense it is the complete form of a coarctation of the aorta. Almost all patients also have other cardiac anomalies, including a ventricular septal defect (VSD), aorto-pulmonary window, and truncus arteriosus. There are three types of interrupted aortic arch, with type B being the most common. Interrupted aortic arch is often associated with DiGeorge syndrome.
A vascular ring is a congenital defect in which there is an abnormal formation of the aorta and/or its surrounding blood vessels. The trachea and esophagus are completely encircled and sometimes compressed by a "ring" formed by these vessels, which can lead to breathing and digestive difficulties.

Aortopulmonary window (APW) is a faulty connection between the aorta and the main pulmonary artery that results in a significant left-to-right shunt. The aortopulmonary window is the rarest of septal defects, accounting for 0.15-0.6% of all congenital heart malformations. An aortopulmonary window can develop alone or in up to 50% of cases alongside other cardiac defects such as interrupted aortic arch, coarctation of the aorta, transposition of great vessels, and tetralogy of Fallot.

Injury of the thoracic aorta refers to any injury which affects the portion of the aorta which lies within the chest cavity. Injuries of the thoracic aorta are usually the result of physical trauma; however, they can also be the result of a pathological process. The main causes of this injury are deceleration and crush injuries. There are different grades to injuries to the aorta depending on the extent of injury, and the treatment whether surgical or medical depends on that grade. It is difficult to determine if a patient has a thoracic injury just by their symptoms, but through imaging and a physical exam the extent of injury can be determined. All patients with a thoracic aortic injury need to be treated either surgically with endovascular repair or open surgical repair or with medicine to keep their blood pressure and heart rate in the appropriate range. However, most patients that have a thoracic aortic injury do not live for 24 hours.

Hypoplastic right heart syndrome (HRHS) is a congenital heart defect in which the structures on the right side of the heart, particularly the right ventricle, are underdeveloped. This defect causes inadequate blood flow to the lungs, and thus a cyanotic infant.

Right-sided aortic arch is a rare anatomical variant in which the aortic arch is on the right side rather than on the left. During normal embryonic development, the aortic arch is formed by the left fourth aortic arch and the left dorsal aorta. In people with a right-sided aortic arch, instead the right dorsal aorta persists and the distal left aorta disappears.

Absent pulmonary valve syndrome(APVS) is a congenital heart defect that occurs when the flaps of the pulmonary valve do not develop or are severely underdeveloped (hypoplasia) resulting in aneurysms (dilation) of the pulmonary arteries and softening of the trachea and bronchi (tracheobronchomalacia). Usually, APVS occurs together with other congenital heart defects, most commonly ventricular septal defect and right ventricular outflow tract obstruction. It is sometimes considered a variant of Tetralogy of Fallot. The first case of absent pulmonary valve syndrome was reported Crampton in 1830.
