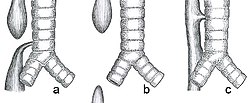
a) Esophageal atresia with distal tracheoesophageal fistula (86%), Gross C. b) Isolated esophageal atresia without tracheoesophageal fistula (7%), Gross A. c) H-type tracheoesophageal fistula (4%), Gross E.[1]
Esophageal atresia is a congenital medical condition (birth defect) that affects the alimentary tract. It causes the esophagus to end in a blind-ended pouch rather than connecting normally to the stomach. It comprises a variety of congenital anatomic defects that are caused by an abnormal embryological development of the esophagus. It is characterized anatomically by a congenital obstruction of the esophagus with interruption of the continuity of the esophageal wall.[2]
The genetic causes of EA/TEF include chromosome anomalies or variants in genes involved in critical developmental processes which are dosage sensitive. Several EA/TEF risk genes have been discovered include the transcriptional regulators SOX2, MYCN, CHD7, FANCB, and members of FOX transcription factor family.[3]
Others plausible candidate genes in the etiology of EA/TEF were identified as APC2, AMER3, PCDH1, GTF3C1, POLR2B, RAB3GAP2, and ITSN1.[4]
Signs and symptoms
Plain X-ray of the chest and abdomen showing a feeding tube unable to move beyond an upper esophageal pouch.Plain x-ray with contrast in the upper esophagus above the atresia.
This birth defect arises in the fourth fetal week, when the trachea and esophagus should begin to separate from each other.[5]
Any attempt at feeding could cause aspiration pneumonia as the milk collects in the blind pouch and overflows into the trachea and lungs. Furthermore, a fistula between the lower esophagus and trachea may allow stomach acid to flow into the lungs and cause damage. Because of these dangers, the condition must be treated as soon as possible after birth resulting in emergency neonatal surgery.[7][8]
Associated birth defects
Other birth defects may co-exist, particularly in the heart, but sometimes also in the anus, spinal column, or kidneys. This is known as VACTERL association because of the involvement of Vertebral column, Anorectal, Cardiac, Tracheal, Esophageal, Renal, and Limbs. It is associated with polyhydramnios in the third trimester.[9]
Diagnosis
This condition may be visible, after about 26 weeks, on an ultrasound. On antenatal USG, the finding of an absent or small stomach in the setting of polyhydramnios was considered a potential symptom of esophageal atresia. However, these findings have a low positive predictive value. The upper neck pouch sign is another sign that helps in the antenatal diagnosis of esophageal atresia and it may be detected soon after birth as the affected infant will be unable to swallow its own saliva.[10]
On plain X-ray, a feeding tube will not be seen passing through the esophagus and remain coiled in the upper oesophageal pouch.[11]
Very rare complete absence of the esophagus, not included in classification by Gross or Ladd
N/A
Type A
Type 2
I
"Long Gap", "Pure" or "Isolated" Esophageal Atresia
Characterized by the presence of a "gap" between the two esophageal blind pouches with no fistula present.
7%
Type B
Type 3A
II
Esophageal Atresia with proximal TEF (tracheoesophageal fistula)
The upper esophageal pouch connects abnormally to the trachea. The lower esophageal pouch ends blindly.
2-3%
Type C
Type 3B
III, IV
Esophageal Atresia with distal TEF (tracheoesophageal fistula)
The lower esophageal pouch connects abnormally to the trachea. The upper esophageal pouch ends blindly.
86%
Type D
Type 3C
V
Esophageal Atresia with both proximal and distal TEFs (two tracheoesophageal fistulas)
Both the upper and lower esophageal pouch make an abnormal connection with the trachea in two separate, isolated places.
<1%
Type E
Type 4
-
TEF (tracheoesophageal fistula) ONLY with no Esophageal Atresia, H-Type
Esophagus fully intact and capable of its normal functions, however, there is an abnormal connection between the esophagus and the trachea. Not included in classification by Ladd
4%
Treatment
Surgical treatment of the condition.Schematic representation.
Treatments for the condition vary depending on its severity. The most immediate and effective treatment in the majority of cases is a surgical repair to close the fistula/s and reconnect the two ends of the esophagus to each other. Although this is usually done through an incision between the ribs on right side of the baby, a technique using three small incisions (thoracoscopy) is being used at some centers.[15]
In a minority of cases, the gap between upper and lower esophageal segments may be too long to bridge. In these situations traditional surgical approaches include gastrostomy followed by gastric pull-up, colonic transposition and jejunum transposition.[16] Gastric pull-up has been the preferred approach at many specialized centers, including Great Ormond Street (London) and Mott Children's Hospital (Ann Arbor).[17]Gastrostomy, or G-tube, allows for tube feedings into the stomach through the abdominal wall. Often a cervical esophagostomy will also be done, to allow the saliva which is swallowed to drain out a hole in the neck. Months or years later, the esophagus may be repaired, sometimes by using a segment of bowel brought up into the chest, interposing between the upper and lower segments of esophagus.[18]
In some of these so-called long gap cases, though, an advanced surgical treatment developed by John Foker, MD,[19] may be utilized to elongate and then join the short esophageal segments. Using the Foker technique, surgeons place traction sutures in the tiny esophageal ends and increase the tension on these sutures daily until the ends are close enough to be sewn together. The result is a normally functioning esophagus, virtually indistinguishable from one congenitally well formed. Unfortunately, the results have been somewhat difficult to replicate by other surgeons and the need for multiple operations has tempered enthusiasm for this approach. The optimal treatment in cases of long gap esophageal atresia remains controversial.[20]
Magnetic compression method is another method for repairing long-gap esophageal atresia. This method does not require replacing the missing section with grafts of the intestine or other body parts. Using electromagnetic force to attract the upper and lower ends of the esophagus together was first tried in the 1970s by using steel pellets attracted to each other by applying external electromagnets to the patient. In the 2000s a further refinement was developed by Mario Zaritzky's group and others.[21] The newer method uses permanent magnets and a balloon.
The magnets are inserted into the upper pouch via the baby's mouth or nose, and the lower via the gastrotomy feeding tube hole (which would have had to be made anyway to feed the baby, therefore not requiring any additional surgery).
The distance between the magnets is controlled by a balloon in the upper pouch, between the end of the pouch and the magnet. This also controls the force between the magnets so it is not strong enough to cause damage.
After the ends of the esophagus have stretched enough to touch, the upper magnet is replaced by one without a balloon and the stronger magnetic attraction causes the ends to fuse (anastomosis).[22][23][24][25]
In April 2015 Annalise Dapo became the first patient in the United States to have their esophageal atresia corrected using magnets.[22][26]
Complications
Postoperative complications may include a leak at the site of closure of the esophagus. Sometimes a stricture, or tight spot, will develop in the esophagus, making it difficult to swallow. Esophageal stricture can usually be dilated using medical instruments. In later life, most children with this disorder will have some trouble with either swallowing or heartburn or both. Esophageal dismotility occurs in 75-100% of patients. After esophageal repair (anastomosis) the relative flaccidity of former proximal pouch (blind pouch, above) along with esophageal dysmotility can cause fluid buildup during feeding. Owing to proximity, pouch ballooning can cause tracheal occlusion. Severe hypoxia ("dying spells") follows and medical intervention can often be required.[citation needed]
Tracheomalacia (a softening of the trachea), usually above the carina (carina of trachea), but sometimes extensive in the lower bronchial tree as well, is another possible serious complication. A variety of treatments for tracheomalacia associated with esophageal atresia are available. If not severe, the condition can be managed expectantly since the trachea will usually stiffen as the infant matures into the first year of life. When only the trachea above the carina is compromised, one of the "simplest" interventions is aortopexy wherein the aortic loop is attached to the rear of the sternum, thereby mechanically relieving pressure from the softened trachea. An even simpler intervention is stenting. However, epithelial cell proliferation and potential incorporation of the stent into the trachea can make subsequent removal dangerous.[citation needed]
The incidence of asthma, bronchitis, bronchial hyperresponsiveness, and recurrent infections in adolescent and adult esophageal atresia survivors far exceeds that of their healthy peers.[27] During the first decade of surgical repair of EA, as much as 20% of patients died from pneumonia. From there on, pneumonia has remained as a major pulmonary complication and a reason for readmissions after repair of EA.[27][28] The risk factors of pneumonia within the first five years of life include other acute respiratory infections and high number of esophageal dilatations.[29]
↑Way, Colin; Wayne, Carolyn; Grandpierre, Viviane; Harrison, Brittany J.; Travis, Nicole; Nasr, Ahmed (2019-11-01). "Thoracoscopy vs. thoracotomy for the repair of esophageal atresia and tracheoesophageal fistula: a systematic review and meta-analysis". Pediatric Surgery International. 35 (11): 1167–1184. doi:10.1007/s00383-019-04527-9. ISSN1437-9813. PMID31359222.
↑Gross RE (1953). The Surgery of Infancy and Childhood. Philadelphia: WB Saunders.
↑Vogt EC (November 1929). "Congenital esophageal atresia". American Journal of Roentgenology. 22: 463–465.
↑Ladd WE (1944). "The surgical treatment of esophageal atresia and tracheoesophageal fistulas". The New England Journal of Medicine. 230 (21): 625–637. doi:10.1056/nejm194405252302101.
↑Kunisaki SM, Foker JE (June 2012). "Surgical advances in the fetus and neonate: esophageal atresia". Clinics in Perinatology. 39 (2): 349–61. doi:10.1016/j.clp.2012.04.007. PMID22682384.
↑Louhimo I, Lindahl H (1983). "Esophageal atresia: primary results of 500 consecutively treated patients". J Pediatr Surg. 18 (3): 217–229. doi:10.1016/s0022-3468(83)80089-x. PMID6875767.
↑Nurminen P, Koivusalo A, Hukkinen M, Pakarinen M (December 2019). "Pneumonia after Repair of Esophageal Atresia-Incidence and Main Risk Factors". European Journal of Pediatric Surgery. 29 (6): 504–509. doi:10.1055/s-0038-1675775. hdl:10138/300624. PMID30469161. S2CID53719974.
Further reading
Harmon CM, Coran AG (1998). "Congenital Anomalies of the Esophagus". Pediatric Surgery (5thed.). St Louis, KY: Elsevier Science Health Science Division. ISBN0-8151-6518-8.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.