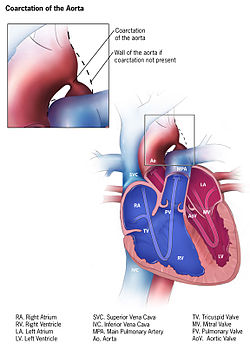
Coarctation of the aorta (CoA)[1][2] is a congenital condition whereby the aorta is narrow, usually in the area where the ductus arteriosus (ligamentum arteriosum after regression) inserts. The word coarctation means "pressing or drawing together; narrowing". Coarctations are most common in the aortic arch. The arch may be small in babies with coarctations. Other heart defects may also occur when coarctation is present, typically occurring on the left side of the heart. When a patient has a coarctation, the left ventricle has to work harder. Since the aorta is narrowed, the left ventricle must generate a much higher pressure than normal in order to force enough blood through the aorta to deliver blood to the lower part of the body. If the narrowing is severe enough, the left ventricle may not be strong enough to push blood through the coarctation, thus resulting in a lack of blood to the lower half of the body. Physiologically its complete form is manifested as interrupted aortic arch.[citation needed]
Preductal coarctation: The narrowing is proximal to the ductus arteriosus. Blood flow to the aorta that is distal to the narrowing is dependent on the ductus arteriosus; therefore severe coarctation can be life-threatening. Preductal coarctation results when an intracardiac anomaly during fetal life decreases blood flow through the left side of the heart, leading to hypoplastic development of the aorta. This is the type seen in approximately 5% of infants with Turner syndrome.[4][5]
Ductal coarctation: The narrowing occurs at the insertion of the ductus arteriosus. This kind usually appears when the ductus arteriosus closes.
Postductal coarctation: The narrowing is distal to the insertion of the ductus arteriosus. Even with an open ductus arteriosus, blood flow to the lower body can be impaired. This type is most common in adults. It is associated with notching of the ribs (because of collateral circulation), hypertension in the upper extremities, and weak pulses in the lower extremities. Postductal coarctation is most likely the result of the extension of a muscular artery (ductus arteriosus) into an elastic artery (aorta) during fetal life, where the contraction and fibrosis of the ductus arteriosus upon birth subsequently narrows the aortic lumen.[6]
Aortic coarctation and aortic stenosis are both forms of aortic narrowing. In terms of word root meanings, the names are not different, but a conventional distinction in their usage allows differentiation of clinical aspects. This spectrum is dichotomized by the idea that aortic coarctation occurs in the aortic arch, at or near the ductus arteriosus, whereas aortic stenosis occurs in the aortic root, at or near the aortic valve.[7] This naturally could present the question of the dividing line between a post valvular stenosis and a preductal coarctation; nonetheless, the dichotomy has a practical use, as most defects are either one or the other.[citation needed]
Signs and symptoms
In mild cases, children may show no signs or symptoms at first and their condition may not be diagnosed until later in life. Some children born with coarctation of the aorta have additional heart defects, such as aortic stenosis, ventricular septal defect, patent ductus arteriosus or mitral valve abnormalities.[citation needed]
Coarctation is about twice as common in boys as it is in girls. It is frequently found in girls who have Turner syndrome.[8]
Symptoms may be absent with mild narrowings (coarctation). When present, they include breathing difficulties, poor appetite or trouble feeding, and failure to thrive. Later on, children may develop symptoms related to problems with blood flow and an enlarged heart. They may experience dizziness or shortness of breath, fainting or near-fainting episodes, chest pain, abnormal tiredness or fatigue, headaches, or nosebleeds. They have cold legs and feet or have pain in their legs with exercise (intermittent claudication).[8]
In cases of more severe coarctations, babies may develop serious problems soon after birth because not enough blood can get through the aorta to the rest of their body. Arterial hypertension in the arms with low blood pressure in the lower extremities is classic. In the lower extremities, weak pulses in the femoral arteries and arteries of the feet are found.[8]
The coarctation typically occurs after the left subclavian artery. However, if situated before it, blood flow to the left arm is compromised and asynchronous or radial pulses of different "strength" may be detected (normal on the right arm, weak or delayed on the left), termed radio-radial delay. In these cases, a difference between the normal radial pulse in the right arm and the delayed femoral pulse in the legs (either side) may be apparent, whilst no such delay would be appreciated with palpation of both delayed left arm and either femoral pulses. On the other hand, a coarctation occurring after the left subclavian artery will produce synchronous radial pulses, but radio-femoral delay will be present under palpation in either arm (both arm pulses are normal compared to the delayed leg pulses).[citation needed]
Diagnosis
With imaging, resorption of the lower part of the ribs may be seen, due to increased blood flow over the neurovascular bundle that runs there. Prestenotic dilatation of the aortic arch and left subclavian artery, as well as indentation at the site of coarctation results in a classic 'figure 3 sign' on x-ray. The characteristic bulging of the sign is caused by dilatation of the aorta due to an indrawing of the aortic wall at the site of cervical rib obstruction, with consequent poststenotic dilatation. This physiology results in the '3' image for which the sign is named.[9][10][11] When the esophagus is filled with barium, a reverse 3 or E sign is often seen and represents a mirror image of the areas of prestenotic and poststenotic dilatation.[12]
Unfortunately, coarctations can not be prevented because they are usually present at birth. The best thing for patients who are affected by coarctations is early detection. Some signs that can lead to a coarctation have been linked to pathologies such as Turner syndrome, bicuspid aortic valve, and other family heart conditions.[5]
Treatment
In adults and children found to have coarctation, treatment is conservative if asymptomatic, but may require surgical resection of the narrow segment if there is arterial hypertension. The first operations to treat coarctation were carried out by Clarence Crafoord in Sweden in 1944.[14] In some cases angioplasty can be performed to dilate the narrowed artery, with or without the placement of a stent graft.[citation needed]
For fetuses at high risk for developing coarctation, a novel experimental treatment approach is being investigated, wherein the mother inhales 45% oxygen three times a day (3 x 3–4 hours) beyond 34 weeks of gestation. The oxygen is transferred via the placenta to the fetus and results in dilatation of the fetal lung vessels. As a consequence, the flow of blood through the fetal circulatory system increases, including that through the underdeveloped arch. In suitable fetuses, marked increases in aortic arch dimensions have been observed over treatment periods of about two to three weeks.[15]
The long-term outcome is very good.[vague] Some patients may, however, develop a narrowing (stenosis) or dilatation at the previous coarctation site. All patients with unrepaired or repaired aortic coarctation require follow-up in specialized Congenital Heart Disease centers.[citation needed]
Complications of surgery
Surgical treatment involves resection of the stenosed segment and re-anastomosis. Two complications specific to this surgery are left recurrent nerve palsy and chylothorax, as the recurrent laryngeal nerve and thoracic duct are in the vicinity. Chylothorax is a troublesome complication and is usually managed conservatively by adjusting the diet to eliminate long-chain fatty acids and supplementing medium-chain triglycerides. When conservative management fails surgical intervention is then most often required.[16] Fluorescein dye can aid in the localisation of chyle leak.[17]
Prognosis
Side effects
Previously, hypertension was defined as a blood pressure of 140/90mm Hg but has since been revised by the American College of Cardiology/American Heart Association Task Force to a blood pressure of 130/80mm Hg or higher in adults.[18] This is a severe problem for the heart and can cause many other complications. In a study of 120 coarctation repair recipients done in Groningen, The Netherlands, twenty-nine patients (25%) experienced hypertension in the later years of life due to the repair. While hypertension has many different factors that lead to this stage of blood pressure, people who have had a coarctation repair — regardless of the age at which the operation was performed — are at much higher risk than the general public of hypertension later in life.[19] Undetected chronic hypertension may result earlier atherosclerosis in the arterial area and can lead to earlier death among coarctation repair patients, at higher rates as time progresses.[19]
Angioplasty is a procedure done to dilate an abnormally narrow section of a blood vessel to allow better blood flow. This is done in a cardiac catheterization laboratory. Typically taking two to three hours, the procedure may take longer but usually patients are able to leave the hospital the same day. After a coarctation repair 20-60% of infant patients may experience reoccurring stenosis at the site of the original operation. This can be fixed by either another coarctectomy[citation needed].[20]
Coronary artery disease (CAD) is a major issue for patients who have undergone a coarctation repair. Many years after the procedure is done, heart disease not only has an increased chance of affecting coarctation patients, but also progresses through the levels of severity at an alarmingly increased rate. In one study, one fourth of the patients who experienced a coarctation later died of heart disease, some at a relatively young age.[21][22]
Clinical criteria are used in most studies when defining recurrence of coarctation (recoarctation) when blood pressure is at a difference of >20 mmHg between the lower and upper limbs. This procedure is most common in infant patients and is uncommon in adult patients. 10.8% of infant patients underwent recoarctations at less than two years of age while another 3.1% of older children received a recoarctation.[23]
People who have had a coarctation of the aorta are likely to have bicuspid aortic valve disease. Between 20% and 85% of patients are affected by this disease. Bicuspid aortic valve disease is a big contributor to cardiac failure, which in turn makes up roughly 20% of late deaths to coarctation patients.[23]
Follow-up
Because of the risk of recoarctation and late hypertension, check-ups are needed once a year or less frequently depending on the individual case. It is important to visit the cardiologist on a regular basis.[19] Depending on the severity of the patient's condition, which is evaluated on a case-by-case level, visiting a cardiologist can be a once a year or less frequent surveillance check-up. Keeping a regular schedule of appointments with a cardiologist after a coarctation procedure is complete helps increase the chances of optimal health for the patients. Nowadays, life expectancy is considered normal given the repair was successfully done in early childhood. Treatment of recoarctation is usually successfully done without the need for open-heart surgery. Recoarctation is increasingly less common in the modern era. Late hypertension does also seem to be much less of a problem if the coarctation repair was performed within the first 5 years of life. Life expectancy and quality of life are therefore the same or very close to that of the normal population, but check ups are recommended so that those few percent who need further treatment get it in time.[24]
History
The condition was largely unidentified until the mid-20th century. History of the condition prior to 1945 has been understood via post-mortem records, the first series of which was published in 1928, which examined cases as far back as 1791. The first surgery for coarctation of the aorta was performed by Clarence Crafoord and G. Nylin on October 19, 1944 in Stockholm, Sweden on a 12-year old boy.[25][26][27]
An anecdotal history statement describes the first diagnosed case of the coarctation of the aorta in Julia the daughter of the French poet Alphonse de Lamartine after the autopsy in 1832 in Beirut, the referenced manuscript still exists in one of the Maronite monasteries in Mount Lebanon.[citation needed]
Gallery
Intraoperative image of aortic coarctation with aneurysmically changed intercostal arteries
Diagram - end-to-end anastomosis
Coarctatio aortae - after excision a narrowing
Coarctatio aortae - after end-to-end anastomosis.
Aortic coarctation using different imaging techniques[28]
↑ Kohl, T; Tchatcheva, K; Stressig, R; Geipel, A; Heitzer, S; Gembruch, U (2008). "Maternal hyperoxygenation in late gestation promotes rapid increase of cardiac dimensions in fetuses with hypoplastic left hearts with intrinsically normal or slightly abnormal aortic and mitral valves". Ultraschall in der Medizin. 29 (S 2). doi:10.1055/s-2008-1080778.
1 2 3 Kuroczynski, Wlodzimierz.; Hartert, Marc; Pruefer, Diethard; Pitzer-Hartert, Katrin; Heinemann, Markus; Vahl, Christian-Friedrich (2008). "Surgical treatment of aortic coarctation in adults: Beneficial effect on arterial hypertension". Cardiol J. 8 (6): 537–542. PMID19039758.
↑ Beekman, Robert H.; Rocchini, Albert P.; Behrendt, Douglas M.; Bove, Edward L.; Dick, Macdonald; Crowley, Dennis C.; Rebecca Snider, A.; Rosenthal, Amnon (1986). "Long-term outcome after repair of coarctation in infancy: Subclavian angioplasty does not reduce the need for reoperation". Journal of the American College of Cardiology. 8 (6): 1406–11. doi:10.1016/s0735-1097(86)80314-x. PMID2946743. S2CID32842425.
↑ Cohen, M.; Fuster, V.; Steele, P. M.; Driscoll, D.; McGoon, D. C. (1989). "Coarctation of the aorta. Long-term follow-up and prediction of outcome after surgical correction". Circulation. 80 (4): 840–5. doi:10.1161/01.CIR.80.4.840. PMID2791247.
Toro-Salazar, Olga H; Steinberger, Julia; Thomas, William; Rocchini, Albert P; Carpenter, Becky; Moller, James H (2002). "Long-term follow-up of patients after coarctation of the aorta repair". The American Journal of Cardiology. 89 (5): 541–7. doi:10.1016/S0002-9149(01)02293-7. PMID11867038.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.