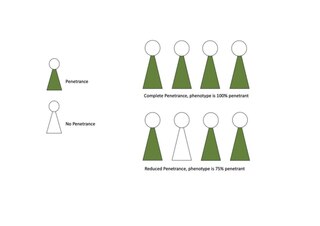
Penetrance in genetics is the proportion of individuals carrying a particular variant of a gene (genotype) that also expresses an associated trait (phenotype). In medical genetics, the penetrance of a disease-causing mutation is the proportion of individuals with the mutation that exhibit clinical symptoms among all individuals with such mutation. For example: If a mutation in the gene responsible for a particular autosomal dominant disorder has 95% penetrance, then 95% of those with the mutation will go on to develop the disease, showing its phenotype, whereas 5% will not.

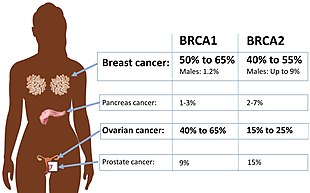
Breast cancer type 1 susceptibility protein is a protein that in humans is encoded by the BRCA1 gene. Orthologs are common in other vertebrate species, whereas invertebrate genomes may encode a more distantly related gene. BRCA1 is a human tumor suppressor gene and is responsible for repairing DNA.

Ovarian cancer is a cancerous tumor of an ovary. It may originate from the ovary itself or more commonly from communicating nearby structures such as fallopian tubes or the inner lining of the abdomen. The ovary is made up of three different cell types including epithelial cells, germ cells, and stromal cells. When these cells become abnormal, they have the ability to divide and form tumors. These cells can also invade or spread to other parts of the body. When this process begins, there may be no or only vague symptoms. Symptoms become more noticeable as the cancer progresses. These symptoms may include bloating, vaginal bleeding, pelvic pain, abdominal swelling, constipation, and loss of appetite, among others. Common areas to which the cancer may spread include the lining of the abdomen, lymph nodes, lungs, and liver.

Fanconi anemia (FA) is a rare, autosomal recessive, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.

BRCA2 and BRCA2 are human genes and their protein products, respectively. The official symbol and the official name are maintained by the HUGO Gene Nomenclature Committee. One alternative symbol, FANCD1, recognizes its association with the FANC protein complex. Orthologs, styled Brca2 and Brca2, are common in other vertebrate species. BRCA2 is a human tumor suppressor gene, found in all humans; its protein, also called by the synonym breast cancer type 2 susceptibility protein, is responsible for repairing DNA.

CHEK2 is a tumor suppressor gene that encodes the protein CHK2, a serine-threonine kinase. CHK2 is involved in DNA repair, cell cycle arrest or apoptosis in response to DNA damage. Mutations to the CHEK2 gene have been linked to a wide range of cancers.
Lethal alleles are alleles that cause the death of the organism that carries them. They are usually a result of mutations in genes that are essential for growth or development. Lethal alleles may be recessive, dominant, or conditional depending on the gene or genes involved.

Fanconi anemia group D2 protein is a protein that in humans is encoded by the FANCD2 gene. The Fanconi anemia complementation group (FANC) currently includes FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN and FANCO. Fanconi anemia proteins, including FANCD2, are an emerging therapeutic target in cancer

Fanconi anemia group J protein is a protein that in humans is encoded by the BRCA1-interacting protein 1 (BRIP1) gene.

Partner and localizer of BRCA2, also known as PALB2 or FANCN, is a protein which in humans is encoded by the PALB2 gene.
Genetic heterogeneity occurs through the production of single or similar phenotypes through different genetic mechanisms. There are two types of genetic heterogeneity: allelic heterogeneity, which occurs when a similar phenotype is produced by different alleles within the same gene; and locus heterogeneity, which occurs when a similar phenotype is produced by mutations at different loci.

PARP inhibitors are a group of pharmacological inhibitors of the enzyme poly ADP ribose polymerase (PARP).

A BRCA mutation is a mutation in either of the BRCA1 and BRCA2 genes, which are tumour suppressor genes. Hundreds of different types of mutations in these genes have been identified, some of which have been determined to be harmful, while others have no proven impact. Harmful mutations in these genes may produce a hereditary breast–ovarian cancer syndrome in affected persons. Only 5–10% of breast cancer cases in women are attributed to BRCA1 and BRCA2 mutations, but the impact on women with the gene mutation is more profound. Women with harmful mutations in either BRCA1 or BRCA2 have a risk of breast cancer that is about five times the normal risk, and a risk of ovarian cancer that is about ten to thirty times normal. The risk of breast and ovarian cancer is higher for women with a high-risk BRCA1 mutation than with a BRCA2 mutation. Having a high-risk mutation does not guarantee that the woman will develop any type of cancer, or imply that any cancer that appears was actually caused by the mutation, rather than some other factor.
FANC proteins are a network of at least 15 proteins that are associated with a cell process known as the Fanconi anemia.
Alan D. D'Andrea is an American cancer researcher and the Fuller American Cancer Society Professor of Radiation Oncology at Harvard Medical School. D'Andrea's research at the Dana Farber Cancer Institute focuses on chromosome instability and cancer susceptibility. He is currently the director of the Center for DNA Damage and Repair and the director of the Susan F. Smith Center for Women's Cancer.
A hereditary cancer syndrome is a genetic disorder in which inherited genetic mutations in one or more genes predispose the affected individuals to the development of cancer and may also cause early onset of these cancers. Hereditary cancer syndromes often show not only a high lifetime risk of developing cancer, but also the development of multiple independent primary tumors.

A variant of uncertainsignificance (VUS) is a genetic variant that has been identified through genetic testing but whose significance to the function or health of an organism is not known. Two related terms are "gene of uncertain significance" (GUS), which refers to a gene that has been identified through genome sequencing but whose connection to a human disease has not been established, and "insignificant mutation", referring to a gene variant that has no impact on the health or function of an organism. The term "variant' is favored in clinical practice over "mutation" because it can be used to describe an allele more precisely. When the variant has no impact on health, it is called a "benign variant". When it is associated with a disease, it is called a "pathogenic variant". A "pharmacogenomic variant" has an effect only when an individual takes a particular drug and therefore is neither benign nor pathogenic.

Prophylactic salpingectomy is a preventative surgical technique performed on patients who are at higher risk of having ovarian cancer, such as individuals who may have pathogenic variants of the BRCA1 or BRCA2 gene. Originally salpingectomy was used in cases of ectopic pregnancies. As a preventative surgery, however, it involves the removal of the fallopian tubes. By not removing the ovaries this procedure is advantageous to individuals who are still of child bearing age. It also reduces risks such as cardiovascular disease and osteoporosis which are associated with the removal of the ovaries.

Breast and ovarian cancer does not necessarily imply that both cancers occur at the same time, but rather that getting one cancer would lead to the development of the other within a few years. Women with a history of breast cancer have a higher chance of developing ovarian cancer, vice versa.

The SEE-FIM protocol is a pathology dissection protocol for Sectioning and Extensively Examining the Fimbria (SEE-FIM). This protocol is intended to provide for the optimal microscopic examination of the distal fallopian tube (fimbria) to identify either cancerous or precancerous conditions in this organ.