Variants in this gene can cause hereditary nonpolyposis colon cancer (Lynch syndrome). It is a human homolog of the E. coli DNA mismatch repair gene, mutL, which mediates protein-protein interactions during mismatch recognition, strand discrimination, and strand removal. Defects in MLH1 are associated with the microsatellite instability observed in hereditary nonpolyposis colon cancer. Alternatively spliced transcript variants encoding different isoforms have been described, but their full-length natures have not been determined.[5]
Role in DNA mismatch repair
MLH1 protein is one component of a system of seven DNA mismatch repair proteins that work coordinately in sequential steps to initiate repair of DNA mismatches in humans.[6] Defects in mismatch repair, found in about 13% of colorectal cancers, are much more frequently due to deficiency of MLH1 than deficiencies of other DNA mismatch repair proteins.[7] The seven DNA mismatch repair proteins in humans are MLH1, MLH3, MSH2, MSH3, MSH6, PMS1 and PMS2.[6] In addition, there are Exo1-dependent and Exo1-independent DNA mismatch repair subpathways.[8]
DNA mismatches occur where one base is improperly paired with another base, or where there is a short addition or deletion in one strand of DNA that is not matched in the other strand. Mismatches commonly occur as a result of DNA replication errors or during genetic recombination. Recognizing those mismatches and repairing them is important for cells because failure to do so results in microsatellite instability] and an elevated spontaneous mutation rate (mutator phenotype). Among 20 cancers evaluated, microsatellite instable colon cancer (mismatch repair deficient) had the second highest frequency of mutations (after melanoma).
A heterodimer between MSH2 and MSH6 first recognizes the mismatch, although a heterodimer between MSH2 and MSH3 also can start the process. The formation of the MSH2-MSH6 heterodimer accommodates a second heterodimer of MLH1 and PMS2, although a heterodimer between MLH1 and either PMS3 or MLH3 can substitute for PMS2. This protein complex formed between the 2 sets of heterodimers enables initiation of repair of the mismatch defect.[6]
Micrograph showing loss of staining for MLH1 in colorectal adenocarcinoma in keeping with DNA mismatch repair (left of image) and benign colorectal mucosa (right of image)
Only a minority of sporadic cancers with a DNA repair deficiency have a mutation in a DNA repair gene. However, a majority of sporadic cancers with a DNA repair deficiency do have one or more epigenetic alterations that reduce or silence DNA repair gene expression.[19] In the table above, the majority of deficiencies of MLH1 were due to methylation of the promoter region of the MLH1 gene. Another epigenetic mechanism reducing MLH1 expression is over-expression of miR-155.[20] MiR-155 targets MLH1 and MSH2 and an inverse correlation between the expression of miR-155 and the expression of MLH1 or MSH2 proteins was found in human colorectal cancer.[20]
Deficiency in field defects
A field defect is an area or "field" of epithelium that has been preconditioned by epigenetic changes and/or mutations so as to predispose it towards development of cancer. As pointed out by Rubin, "The vast majority of studies in cancer research has been done on well-defined tumors in vivo, or on discrete neoplastic foci in vitro.[21] Yet there is evidence that more than 80% of the somatic mutations found in mutator phenotype human colorectal tumors occur before the onset of terminal clonal expansion."[22] Similarly, Vogelstein et al.[23] point out that more than half of somatic mutations identified in tumors occurred in a pre-neoplastic phase (in a field defect), during growth of apparently normal cells.
In the Table above, MLH1 deficiencies were noted in the field defects (histologically normal tissues) surrounding most of the cancers. If MLH1 is epigenetically reduced or silenced, it would not likely confer a selective advantage upon a stem cell. However, reduced or absent expression of MLH1 would cause increased rates of mutation, and one or more of the mutated genes may provide the cell with a selective advantage. The expression-deficient MLH1 gene could then be carried along as a selectively neutral or only slightly deleterious passenger (hitch-hiker) gene when the mutated stem cell generates an expanded clone. The continued presence of a clone with an epigenetically repressed MLH1 would continue to generate further mutations, some of which could produce a tumor.
Repression in coordination with other DNA repair genes
In a cancer, multiple DNA repair genes are often found to be simultaneously repressed.[19] In one example, involving MLH1, Jiang et al.[24] conducted a study where they evaluated the mRNA expression of 27 DNA repair genes in 40 astrocytomas compared to normal brain tissues from non-astrocytoma individuals. Among the 27 DNA repair genes evaluated, 13 DNA repair genes, MLH1, MLH3, MGMT,NTHL1,OGG1,SMUG1,ERCC1,ERCC2, ERCC3, ERCC4, RAD50, XRCC4 and XRCC5 were all significantly down-regulated in all three grades (II, III and IV) of astrocytomas. The repression of these 13 genes in lower grade as well as in higher grade astrocytomas suggested that they may be important in early as well as in later stages of astrocytoma. In another example, Kitajima et al.[25] found that immunoreactivity for MLH1 and MGMT expression was closely correlated in 135 specimens of gastric cancer and loss of MLH1 and MGMTappeared to be synchronously accelerated during tumor progression.
Deficient expression of multiple DNA repair genes are often found in cancers,[19] and may contribute to the thousands of mutations usually found in cancers (see Mutation frequencies in cancers).
Meiosis
In addition to its role in DNA mismatch repair, MLH1 protein is also involved in meioticcrossing over.[26] MLH1 forms a heterodimer with MLH3 that appears to be necessary for oocytes to progress through metaphase II of meiosis.[27] Female and male MLH1(-/-) mutant mice are infertile, and sterility is associated with a reduced level of chiasmata.[26][28] During spermatogenesis in MLH1(-/-) mutant mice chromosomes often separate prematurely and there is frequent arrest in the first division of meiosis.[26] In humans, a common variant of the MLH1 gene is associated with increased risk of sperm damage and male infertility.[29]
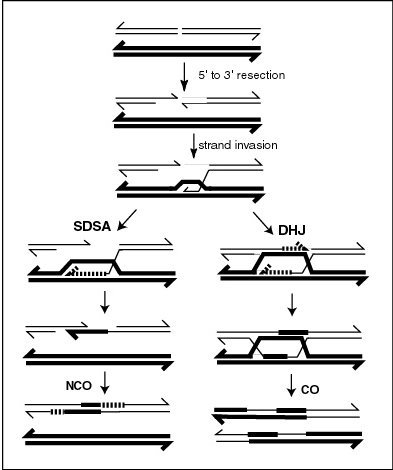
A current model of meiotic recombination, initiated by a double-strand break or gap, followed by pairing with an homologous chromosome and strand invasion to initiate the recombinational repair process. Repair of the gap can lead to crossover (CO) or non-crossover (NCO) of the flanking regions. CO recombination is thought to occur by the Double Holliday Junction (DHJ) model, illustrated on the right, above. NCO recombinants are thought to occur primarily by the Synthesis Dependent Strand Annealing (SDSA) model, illustrated on the left, above. Most recombination events appear to be the SDSA type.
MLH1 protein appears to localize to sites of crossing over in meiotic chromosomes.[26]Recombination during meiosis is often initiated by a DNA double-strand break (DSB) as illustrated in the accompanying diagram. During recombination, sections of DNA at the 5' ends of the break are cut away in a process called resection. In the strand invasion step that follows, an overhanging 3' end of the broken DNA molecule then "invades" the DNA of an homologous chromosome that is not broken forming a displacement loop (D-loop). After strand invasion, the further sequence of events may follow either of two main pathways leading to a crossover (CO) or a non-crossover (NCO) recombinant (see Genetic recombination). The pathway leading to a CO involves a double Holliday junction (DHJ) intermediate. Holliday junctions need to be resolved for CO recombination to be completed.
In the budding yeast Saccharomyces cerevisiae, as in the mouse, MLH1 forms a heterodimer with MLH3. Meiotic CO requires resolution of Holliday junctions through actions of the MLH1-MLH3 heterodimer. The MLH1-MLH3 heterodimer is an endonuclease that makes single-strand breaks in supercoiled double-stranded DNA.[30][31] MLH1-MLH3 binds specifically to Holliday junctions and may act as part of a larger complex to process Holliday junctions during meiosis.[30] MLH1-MLH3 heterodimer (MutL gamma) together with EXO1 and Sgs1 (ortholog of Bloom syndrome helicase) define a joint molecule resolution pathway that produces the majority of crossovers in budding yeast and, by inference, in mammals.[32]
↑ Tawfik HM, El-Maqsoud NM, Hak BH, El-Sherbiny YM (2011). "Head and neck squamous cell carcinoma: mismatch repair immunohistochemistry and promoter hypermethylation of hMLH1 gene". Am J Otolaryngol. 32 (6): 528–36. doi:10.1016/j.amjoto.2010.11.005. PMID21353335.
↑ Zuo C, Zhang H, Spencer HJ, Vural E, Suen JY, Schichman SA, Smoller BR, Kokoska MS, Fan CY (2009). "Increased microsatellite instability and epigenetic inactivation of the hMLH1 gene in head and neck squamous cell carcinoma". Otolaryngol Head Neck Surg. 141 (4): 484–90. doi:10.1016/j.otohns.2009.07.007. PMID19786217. S2CID8357370.
↑ Rubin H (March 2011). "Fields and field cancerization: the preneoplastic origins of cancer: asymptomatic hyperplastic fields are precursors of neoplasia, and their progression to tumors can be tracked by saturation density in culture". BioEssays. 33 (3): 224–31. doi:10.1002/bies.201000067. PMID21254148. S2CID44981539.
↑ Jiang Z, Hu J, Li X, Jiang Y, Zhou W, Lu D (2006). "Expression analyses of 27 DNA repair genes in astrocytoma by TaqMan low-density array". Neurosci. Lett. 409 (2): 112–7. doi:10.1016/j.neulet.2006.09.038. PMID17034947. S2CID54278905.
Paraf F, Sasseville D, Watters AK, Narod S, Ginsburg O, Shibata H, Jothy S (1995). "Clinicopathological relevance of the association between gastrointestinal and sebaceous neoplasms: the Muir-Torre syndrome". Hum. Pathol. 26 (4): 422–7. doi:10.1016/0046-8177(95)90144-2. PMID7705822.
Kolodner RD (1996). "Mismatch repair: mechanisms and relationship to cancer susceptibility". Trends Biochem. Sci. 20 (10): 397–401. doi:10.1016/S0968-0004(00)89087-8. PMID8533151.
Warusavitarne J, Schnitzler M (2007). "The role of chemotherapy in microsatellite unstable (MSI-H) colorectal cancer". International Journal of Colorectal Disease. 22 (7): 739–48. doi:10.1007/s00384-006-0228-0. PMID17109103. S2CID6460105.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.