Chromosomal crossover, or crossing over, is the exchange of genetic material during sexual reproduction between two homologous chromosomes' non-sister chromatids that results in recombinant chromosomes. It is one of the final phases of genetic recombination, which occurs in the pachytene stage of prophase I of meiosis during a process called synapsis. Synapsis begins before the synaptonemal complex develops and is not completed until near the end of prophase I. Crossover usually occurs when matching regions on matching chromosomes break and then reconnect to the other chromosome.

Fanconi anemia (FA) is a rare, autosomal recessive, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.
RecQ helicase is a family of helicase enzymes initially found in Escherichia coli that has been shown to be important in genome maintenance. They function through catalyzing the reaction ATP + H2O → ADP + P and thus driving the unwinding of paired DNA and translocating in the 3' to 5' direction. These enzymes can also drive the reaction NTP + H2O → NDP + P to drive the unwinding of either DNA or RNA.

BRCA2 and BRCA2 are human genes and their protein products, respectively. The official symbol and the official name are maintained by the HUGO Gene Nomenclature Committee. One alternative symbol, FANCD1, recognizes its association with the FANC protein complex. Orthologs, styled Brca2 and Brca2, are common in other vertebrate species. BRCA2 is a human tumor suppressor gene, found in all humans; its protein, also called by the synonym breast cancer type 2 susceptibility protein, is responsible for repairing DNA.
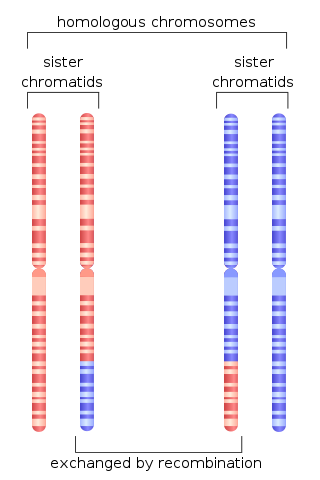
Homologous recombination is a type of genetic recombination in which genetic information is exchanged between two similar or identical molecules of double-stranded or single-stranded nucleic acids.

Bloom syndrome protein is a protein that in humans is encoded by the BLM gene and is not expressed in Bloom syndrome.

Fanconi anemia group C protein is a protein that in humans is encoded by the FANCC gene. This protein delays the onset of apoptosis and promotes homologous recombination repair of damaged DNA. Mutations in this gene result in Fanconi anemia, a human rare disorder characterized by cancer susceptibility and cellular sensitivity to DNA crosslinks and other damages.

Fanconi anaemia, complementation group A, also known as FAA, FACA and FANCA, is a protein which in humans is encoded by the FANCA gene. It belongs to the Fanconi anaemia complementation group (FANC) family of genes of which 12 complementation groups are currently recognized and is hypothesised to operate as a post-replication repair or a cell cycle checkpoint. FANCA proteins are involved in inter-strand DNA cross-link repair and in the maintenance of normal chromosome stability that regulates the differentiation of haematopoietic stem cells into mature blood cells.

Fanconi anemia group D2 protein is a protein that in humans is encoded by the FANCD2 gene. The Fanconi anemia complementation group (FANC) currently includes FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN and FANCO.
Fanconi anemia group G protein is a protein that in humans is encoded by the FANCG gene.

Exonuclease 1 is an enzyme that in humans is encoded by the EXO1 gene.

Fanconi anemia group J protein is a protein that in humans is encoded by the BRCA1-interacting protein 1 (BRIP1) gene.
DNA topoisomerase 3-alpha is an enzyme that in humans is encoded by the TOP3A gene.

E3 ubiquitin-protein ligase FANCL is an enzyme that in humans is encoded by the FANCL gene.

Fanconi anemia, complementation group I (FANCI) also known as KIAA1794, is a protein which in humans is encoded by the FANCI gene. Mutations in the FANCI gene are known to cause Fanconi anemia.

Partner and localizer of BRCA2, also known as PALB2 or FANCN, is a protein which in humans is encoded by the PALB2 gene.

RecQ-mediated genome instability protein 1 is a protein that in humans is encoded by the RMI1 gene.
FANC proteins are a network of at least 15 proteins that are associated with a cell process known as the Fanconi anemia.

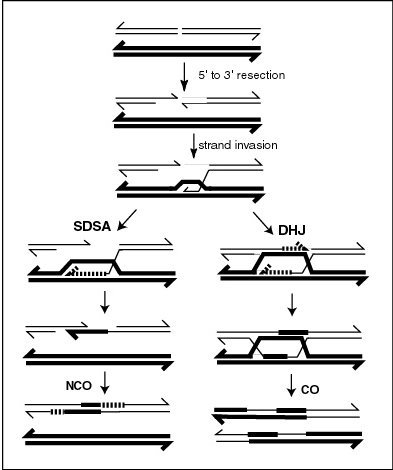
Synthesis-dependent strand annealing (SDSA) is a major mechanism of homology-directed repair of DNA double-strand breaks (DSBs). Although many of the features of SDSA were first suggested in 1976, the double-Holliday junction model proposed in 1983 was favored by many researchers. In 1994, studies of double-strand gap repair in Drosophila were found to be incompatible with the double-Holliday junction model, leading researchers to propose a model they called synthesis-dependent strand annealing. Subsequent studies of meiotic recombination in S. cerevisiae found that non-crossover products appear earlier than double-Holliday junctions or crossover products, challenging the previous notion that both crossover and non-crossover products are produced by double-Holliday junctions and leading the authors to propose that non-crossover products are generated through SDSA.

A double-strand break repair model refers to the various models of pathways that cells undertake to repair double strand-breaks (DSB). DSB repair is an important cellular process, as the accumulation of unrepaired DSB could lead to chromosomal rearrangements, tumorigenesis or even cell death. In human cells, there are two main DSB repair mechanisms: Homologous recombination (HR) and non-homologous end joining (NHEJ). HR relies on undamaged template DNA as reference to repair the DSB, resulting in the restoration of the original sequence. NHEJ modifies and ligates the damaged ends regardless of homology. In terms of DSB repair pathway choice, most mammalian cells appear to favor NHEJ rather than HR. This is because the employment of HR may lead to gene deletion or amplification in cells which contains repetitive sequences. In terms of repair models in the cell cycle, HR is only possible during the S and G2 phases, while NHEJ can occur throughout whole process. These repair pathways are all regulated by the overarching DNA damage response mechanism. Besides HR and NHEJ, there are also other repair models which exists in cells. Some are categorized under HR, such as synthesis-dependent strain annealing, break-induced replication, and single-strand annealing; while others are an entirely alternate repair model, namely, the pathway microhomology-mediated end joining (MMEJ).