In contrast to phosphonium ylides used in the Wittig reaction, phosphonate-stabilized carbanions are more nucleophilic but less basic. Likewise, phosphonate-stabilized carbanions can be alkylated. Unlike phosphonium ylides, the dialkylphosphate salt byproduct is easily removed by aqueousextraction.
Deprotonation by base (B-) to generate the phosphonate carbanion
The Horner–Wadsworth–Emmons reaction begins with the deprotonation of the phosphonate to give the phosphonate carbanion1. Nucleophilic addition of the carbanion onto the aldehyde 2 (or ketone) producing 3a or 3b is the rate-limiting step.[12] If R2 = H, then intermediates 3a and 4a and intermediates 3b and 4b can interconvert with each other.[13] The final elimination of oxaphosphetanes4a and 4b yield (E)-alkene 5 and (Z)-alkene 6, with the by-product being a dialkyl-phosphate.
The mechanism of the Horner-Wadsworth-Emmons reaction
The ratio of alkene isomers5 and 6 is not dependent upon the stereochemical outcome of the initial carbanion addition and upon the ability of the intermediates to equilibrate.
The electron-withdrawing group (EWG) alpha to the phosphonate is necessary for the final elimination to occur. In the absence of an electron-withdrawing group, the final product is the β-hydroxyphosphonate 3a and 3b.[14] However, these β-hydroxyphosphonates can be transformed to alkenes by reaction with diisopropylcarbodiimide.[15]
Stereoselectivity
The Horner–Wadsworth–Emmons reaction favours the formation of (E)-alkenes. In general, the more equilibration amongst intermediates, the higher the selectivity for (E)-alkene formation.
Disubstituted alkenes
Thompson and Heathcock have performed a systematic study of the reaction of methyl 2-(dimethoxyphosphoryl)acetate with various aldehydes.[16] While each effect was small, they had a cumulative effect making it possible to modify the stereochemical outcome without modifying the structure of the phosphonate. They found greater (E)-stereoselectivity with the following conditions:
In a separate study, it was found that bulky phosphonate and bulky electron-withdrawing groups enhance E-alkene selectivity.
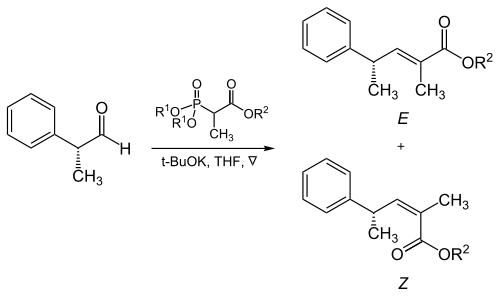
Trisubstituted alkenes
The steric bulk of the phosphonate and electron-withdrawing groups plays a critical role in the reaction of α-branched phosphonates with aliphatic aldehydes.[17]
Example of the Horner–Wadsworth–Emmons reaction with branched phosphonates
Aromatic aldehydes produce almost exclusively (E)-alkenes. In case (Z)-alkenes from aromatic aldehydes are needed, the Still–Gennari modification (see below) can be used.
Olefination of ketones
The stereoselectivity of the Horner–Wadsworth–Emmons reaction of ketones is poor to modest.
W. Clark Still and C. Gennari have developed conditions that give Z-alkenes with excellent stereoselectivity.[23][24] Using phosphonates with electron-withdrawing groups (trifluoroethyl[25]) together with strongly dissociating conditions (KHMDS and 18-crown-6 in THF) nearly exclusive Z-alkene production can be achieved.
The Still modification of the Horner–Wadsworth–Emmons reaction
Ando has suggested that the use of electron-deficient phosphonates accelerates the elimination of the oxaphosphetane intermediates.[26]
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.