Related Research Articles

Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics. It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles. LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.

Titin is a protein that in humans is encoded by the TTN gene. Titin is a protein, over 1 µm in length, that functions as a molecular spring that is responsible for the passive elasticity of muscle. It comprises 244 individually folded protein domains connected by unstructured peptide sequences. These domains unfold when the protein is stretched and refold when the tension is removed.

Mitochondrial myopathies are types of myopathies associated with mitochondrial disease. Adenosine triphosphate (ATP), the chemical used to provide energy for the cell, cannot be produced sufficiently by oxidative phosphorylation when the mitochondrion is either damaged or missing necessary enzymes or transport proteins. With ATP production deficient in mitochondria, there is an over-reliance on anaerobic glycolysis which leads to lactic acidosis either at rest or exercise-induced.

A calpain is a protein belonging to the family of calcium-dependent, non-lysosomal cysteine proteases expressed ubiquitously in mammals and many other organisms. Calpains constitute the C2 family of protease clan CA in the MEROPS database. The calpain proteolytic system includes the calpain proteases, the small regulatory subunit CAPNS1, also known as CAPN4, and the endogenous calpain-specific inhibitor, calpastatin.

Dysferlin also known as dystrophy-associated fer-1-like protein is a protein that in humans is encoded by the DYSF gene. Dysferlin is linked with plasma membrane repair., stabilization of calcium signaling and the development of the T-tubule system of the muscle A defect in the DYSF gene, located on chromosome 2p12-14, results in several types of muscular dystrophy; including Miyoshi myopathy (MM), Limb-girdle muscular dystrophy type 2B (LGMD2B) and Distal Myopathy (DM). A reduction or absence of dysferlin, termed dysferlinopathy, usually becomes apparent in the third or fourth decade of life and is characterised by weakness and wasting of various voluntary skeletal muscles. Pathogenic mutations leading to dysferlinopathy can occur throughout the DYSF gene.

Fukutin is a eukaryotic protein necessary for the maintenance of muscle integrity, cortical histogenesis, and normal ocular development. Mutations in the fukutin gene have been shown to result in Fukuyama congenital muscular dystrophy (FCMD) characterised by brain malformation - one of the most common autosomal-recessive disorders in Japan. In humans this protein is encoded by the FCMD gene, located on chromosome 9q31. Human fukutin exhibits a length of 461 amino acids and a predicted molecular mass of 53.7 kDa.

Calpain-10 is a protein that in humans is encoded by the CAPN10 gene.

Calpain-2 catalytic subunit is a protein that in humans is encoded by the CAPN2 gene.

Myotonin-protein kinase (MT-PK) also known as myotonic dystrophy protein kinase (MDPK) or dystrophia myotonica protein kinase (DMPK) is an enzyme that in humans is encoded by the DMPK gene.

Calpain-1 catalytic subunit(CANP 1) is a protein that in humans is encoded by the CAPN1 gene.
Calpain small subunit 1 (CSS1) is a protein that in humans is encoded by the CAPNS1 gene.

Delta-sarcoglycan is a protein that in humans is encoded by the SGCD gene.

Alpha-sarcoglycan is a protein that in humans is encoded by the SGCA gene.
Myotilin is a protein that in humans is encoded by the MYOT gene. Myotilin also known as TTID is a muscle protein that is found within the Z-disc of sarcomeres.

Gamma-sarcoglycan is a protein that in humans is encoded by the SGCG gene. The α to δ-sarcoglycans are expressed predominantly (β) or exclusively in striated muscle. A mutation in any of the sarcoglycan genes may lead to a secondary deficiency of the other sarcoglycan proteins, presumably due to destabilisation of the sarcoglycan complex. The disease-causing mutations in the α to δ genes cause disruptions within the dystrophin-associated protein (DAP) complex in the muscle cell membrane. The transmembrane components of the DAP complex link the cytoskeleton to the extracellular matrix in adult muscle fibres, and are essential for the preservation of the integrity of the muscle cell membrane.

Protein O-mannosyl-transferase 1 is an enzyme that in humans is encoded by the POMT1 gene. It is a member of the dolichyl-phosphate-mannose-protein mannosyltransferases.
Tripartite motif-containing protein 32 is a protein that in humans is encoded by the TRIM32 gene. Since its discovery in 1995, TRIM32 has been shown to be implicated in a number of diverse biological pathways.

Calpain-9 is a protein that in humans is encoded by the CAPN9 gene.

Anoctamin 5 (ANO5) is a protein that in humans is encoded by the ANO5 gene.
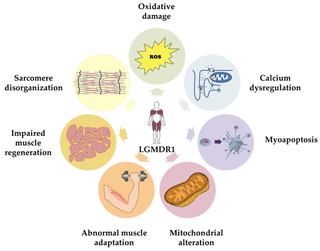
Calpainopathy is the most common type of autosomal recessive limb-girdle muscular dystrophy (LGMD). It preferentially affects the muscles of the hip girdle and shoulder girdle.
References
- ↑ Sorimachi H, Imajoh-Ohmi S, Emori Y, Kawasaki H, Ohno S, Minami Y, Suzuki K (December 1989). "Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-types. Specific expression of the mRNA in skeletal muscle". J. Biol. Chem. 264 (33): 20106–11. doi: 10.1016/S0021-9258(19)47225-6 . PMID 2555341.
- ↑ Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C, Pasturaud P, Roudaut C (May 1995). "Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A". Cell. 81 (1): 27–40. doi: 10.1016/0092-8674(95)90368-2 . PMID 7720071.
- ↑ "Entrez Gene: CAPN3 calpain 3, (p94)".
- ↑ Hoek KS, Schlegel NC, Eichhoff OM, Widmer DS, Praetorius C, Einarsson SO, Valgeirsdottir S, Bergsteinsdottir K, Schepsky A, Dummer R, Steingrimsson E (2008). "Novel MITF targets identified using a two-step DNA microarray strategy". Pigment Cell Melanoma Res. 21 (6): 665–76. doi: 10.1111/j.1755-148X.2008.00505.x . PMID 19067971.
- ↑ Ono Y, Shimada H, Sorimachi H, Richard I, Saido TC, Beckmann JS, Ishiura S, Suzuki K (July 1998). "Functional defects of a muscle-specific calpain, p94, caused by mutations associated with limb-girdle muscular dystrophy type 2A". J. Biol. Chem. 273 (27): 17073–8. doi: 10.1074/jbc.273.27.17073 . PMID 9642272.
- ↑ Sorimachi H, Kinbara K, Kimura S, Takahashi M, Ishiura S, Sasagawa N, Sorimachi N, Shimada H, Tagawa K, Maruyama K (December 1995). "Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence". J. Biol. Chem. 270 (52): 31158–62. doi: 10.1074/jbc.270.52.31158 . PMID 8537379.