
Metabolic syndrome is a clustering of at least three of the following five medical conditions: abdominal obesity, high blood pressure, high blood sugar, high serum triglycerides, and low serum high-density lipoprotein (HDL).
Insulin resistance (IR) is a pathological condition in which cells fail to respond normally to the hormone insulin.
Lipodystrophy syndromes are a group of genetic or acquired disorders in which the body is unable to produce and maintain healthy fat tissue. The medical condition is characterized by abnormal or degenerative conditions of the body's adipose tissue. A more specific term, lipoatrophy, is used when describing the loss of fat from one area. This condition is also characterized by a lack of circulating leptin which may lead to osteosclerosis. The absence of fat tissue is associated with insulin resistance, hypertriglyceridemia, non-alcoholic fatty liver disease (NAFLD) and metabolic syndrome.

A benign tumor is a mass of cells (tumor) that lacks the ability to either invade neighboring tissue or metastasize. When removed, benign tumors usually do not grow back, whereas malignant tumors are cancerous and sometimes do. Unlike most benign tumors elsewhere in the body, benign brain tumors can be life-threatening. Benign tumors generally have a slower growth rate than malignant tumors and the tumor cells are usually more differentiated. They are typically surrounded by an outer surface or stay contained within the epithelium. Common examples of benign tumors include moles and uterine fibroids.
The liver X receptor (LXR) is a member of the nuclear receptor family of transcription factors and is closely related to nuclear receptors such as the PPARs, FXR and RXR. Liver X receptors (LXRs) are important regulators of cholesterol, fatty acid, and glucose homeostasis. LXRs were earlier classified as orphan nuclear receptors, however, upon discovery of endogenous oxysterols as ligands they were subsequently deorphanized.

Laminopathies are a group of rare genetic disorders caused by mutations in genes encoding proteins of the nuclear lamina. They are included in the more generic term nuclear envelopathies that was coined in 2000 for diseases associated with defects of the nuclear envelope. Since the first reports of laminopathies in the late 1990s, increased research efforts have started to uncover the vital role of nuclear envelope proteins in cell and tissue integrity in animals.

Pre-lamin A/C or lamin A/C is a protein that in humans is encoded by the LMNA gene. Lamin A/C belongs to the lamin family of proteins.

Hepatic lipase (HL), also called hepatic triglyceride lipase (HTGL) or LIPC, is a form of lipase, catalyzing the hydrolysis of triacylglyceride. Hepatic lipase is coded by chromosome 15 and its gene is also often referred to as HTGL or LIPC. Hepatic lipase is expressed mainly in liver cells, known as hepatocytes, and endothelial cells of the liver. The hepatic lipase can either remain attached to the liver or can unbind from the liver endothelial cells and is free to enter the body's circulation system. When bound on the endothelial cells of the liver, it is often found bound to HSPG, heparan sulfate proteoglycans (HSPG), keeping HL inactive and unable to bind to HDL or IDL. When it is free in the bloodstream, however, it is found associated with HDL to maintain it inactive. This is because the triacylglycerides in HDL serve as a substrate, but the lipoprotein contains proteins around the triacylglycerides that can prevent the triacylglycerides from being broken down by HL.
Barraquer–Simons syndrome is a rare form of lipodystrophy, which usually first affects the head, and then spreads to the thorax. It is named for Luis Barraquer Roviralta (1855–1928), a Spanish physician, and Arthur Simons (1879–1942), a German physician. Some evidence links it to LMNB2.
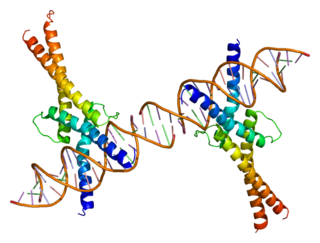
Sterol regulatory element-binding transcription factor 1 (SREBF1) also known as sterol regulatory element-binding protein 1 (SREBP-1) is a protein that in humans is encoded by the SREBF1 gene.

Fatty acid-binding protein 2 (FABP2) also known as Intestinal-type fatty acid-binding protein (I-FABP) is a protein that in humans is encoded by the FABP2 gene.

Lipin-1 is a protein that in humans is encoded by the LPIN1 gene.

Carbohydrate-responsive element-binding protein (ChREBP) also known as MLX-interacting protein-like (MLXIPL) is a protein that in humans is encoded by the MLXIPL gene. The protein name derives from the protein's interaction with carbohydrate response element sequences of DNA.
Congenital generalized lipodystrophy is an extremely rare autosomal recessive condition, characterized by an extreme scarcity of fat in the subcutaneous tissues. It is a type of lipodystophy disorder where the magnitude of fat loss determines the severity of metabolic complications. Only 250 cases of the condition have been reported, and it is estimated that it occurs in 1 in 10 million people worldwide.
Acquired generalized lipodystrophy is a rare skin condition that appears during childhood or adolescence, characterized by fat loss affecting large areas of the body, particularly the face, arms, and legs. There are 4 types of lipodystrophy based on its onset and areas affected: acquired or inherited, and generalized or partial. Both acquired or inherited lipodystrophy present as loss of adipose tissues. The near-total loss of subcutaneous adipose tissue is termed generalized lipodystrophy while the selective loss of adipose tissues is denoted as partial lipodystrophy. Thus, as the name suggests, AGL is a near-total deficiency of adipose tissues in the body that is developed later in life. It is an extremely rare disease that only about 100 cases are reported worldwide. There are three main etiologies of AGL suspected: autoimmune, panniculitis-associated, or idiopathic. After its onset, the disease progresses over a few days, weeks, months, or even in years. Clinical presentations of AGL are similar to other lipodystrophies, including metabolic complications and hypoleptinemia. Treatments are also similar and mainly supportive for symptomatic alleviation. Although HIV- or drug-induced lipodystrophy are a type of acquired lipodystrophy, its origin is very specific and distinct hence is usually not discussed with AGL.
Familial partial lipodystrophy, also known as Köbberling–Dunnigan syndrome, is a rare genetic metabolic condition characterized by the loss of subcutaneous fat.

Wolcott–Rallison syndrome,WRS, is a rare, autosomal recessive disorder with infancy-onset diabetes mellitus, multiple epiphyseal dysplasia, osteopenia, mental retardation or developmental delay, and hepatic and renal dysfunction as main clinical findings. Patients with WRS have mutations in the EIF2AK3 gene, which encodes the eukaryotic translation initiation factor 2-alpha kinase 3. Other disease names include multiple epiphyseal dysplasia and early-onset diabetes mellitus. Most patients with this disease do not survive to adulthood. The majority of WRS patients die from fulminant hepatitis during childhood. There are few reported cases for this disease. Of the 54 families worldwide with reported WRS cases, 22.2% of them are from the Kingdom of Saudi Arabia. Of the 23 WRS patients in Saudi Arabia, all but one is the result of consanguineous marriages. Another country where WRS cases have been found is Kosovo. Here, the Albanian population is also known for consanguineous marriages, but there were some cases involving patients from non-consanguineous parents that were carriers for the same mutant allele.

Familial hypertriglyceridemia is a genetic disorder characterized by the liver overproducing very-low-density lipoproteins (VLDL). As a result, an afflicted individual will have an excessive number of VLDL and triglycerides on a lipid profile. This genetic disorder usually follows an autosomal dominant inheritance pattern. The disorder presents clinically in patients with mild to moderate elevations in triglyceride levels. Familial hypertriglyceridemia is typically associated with other co-morbid conditions such as hypertension, obesity, and hyperglycemia. Individuals with the disorder are mostly heterozygous in an inactivating mutation of the gene encoding for lipoprotein lipase (LPL). This sole mutation can markedly elevate serum triglyceride levels. However, when combined with other medications or pathologies it can further elevate serum triglyceride levels to pathologic levels. Substantial increases in serum triglyceride levels can lead to certain clinical signs and the development of acute pancreatitis.
Seipin is a homo-oligomeric integral membrane protein in the endoplasmic reticulum (ER) that concentrates at junctions with cytoplasmic lipid droplets (LDs). Alternatively, seipin can be referred to as Bernardinelli-Seip congenital lipodystrophy type 2 protein (BSCL2), and it is encoded by the corresponding gene of the same name, i.e. BSCL2. At protein level, seipin is expressed in cortical neurons in the frontal lobes, as well as motor neurons in the spinal cord. It is highly expressed in areas like the brain, testis and adipose tissue. Seipin's function is still unclear but it has been localized close to lipid droplets, and cells knocked out in seipin which have anomalous droplets. Hence, recent evidence suggests that seipin plays a crucial role in lipid droplet biogenesis.
Asprosin is a protein hormone produced by mammals in tissues that stimulates the liver to release glucose into the blood stream. Asprosin is encoded by the gene FBN1 as part of the protein profibrillin and is released from the C-terminus of the latter by specific proteolysis. In the liver, asprosin activates rapid glucose release via a cyclic adenosine monophosphate (cAMP)-dependent pathway.
