
A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosomal abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Ehlers–Danlos syndromes are a group of rare genetic connective tissue disorders. Symptoms may include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. These can be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis.

Weissenbacher–Zweymuller syndrome (WZS), also called Pierre-Robin syndrome with fetal chondrodysplasia, is an autosomal recessive congenital disorder, linked to mutations in the COL11A2 gene, which codes for the α2 strand of collagen type XI. It is a collagenopathy, types II and XI disorder.

Osteogenesis imperfecta (OI), also known as brittle bone disease, is a group of genetic disorders that mainly affect the bones. It results in bones that break easily. The severity may be mild to severe. Other symptoms may include a blue tinge to the whites of the eye, short height, loose joints, hearing loss, breathing problems and problems with the teeth. Complications may include cervical artery dissection and aortic dissection.
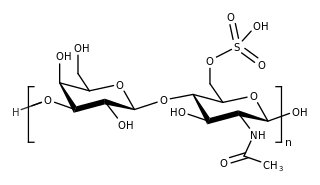
Morquio syndrome, also known as Mucopolysaccharidosis Type IV , is a rare metabolic disorder in which the body cannot process certain types of sugar molecules called glycosaminoglycans. In Morquio syndrome, the specific GAG which builds up in the body is called keratan sulfate. This birth defect, which is autosomal recessive, is a type of lysosomal storage disorder. The buildup of GAGs in different parts of the body causes symptoms in many different organ systems. In the US, the incidence rate for Morquio syndrome is estimated at between 1 in 200,000 and 1 in 300,000 live births.

Hypermobility, also known as double-jointedness, describes joints that stretch farther than normal. For example, some hypermobile people can bend their thumbs backwards to their wrists, bend their knee joints backwards, put their leg behind the head or perform other contortionist "tricks." It can affect one or more joints throughout the body.

Simpson–Golabi–Behmel syndrome (SGBS), is a rare inherited congenital disorder that can cause craniofacial, skeletal, cardiac, and renal abnormalities. The syndrome is inherited in an X-linked recessive fashion, where males express the phenotype and females usually do not. Females that possess one copy of the mutation are considered to be carriers of the syndrome and may express varying degrees of the phenotype.

Robinow syndrome is an extremely rare genetic disorder characterized by short-limbed dwarfism, abnormalities in the head, face, and external genitalia, as well as vertebral segmentation. The disorder was first described in 1969 by human geneticist Meinhard Robinow, along with physicians Frederic N. Silverman and Hugo D. Smith, in the American Journal of Diseases of Children. By 2002, over 100 cases had been documented and introduced into medical literature.
Rabson–Mendenhall syndrome is a rare autosomal recessive disorder characterized by severe insulin resistance. The disorder is caused by mutations in the insulin receptor gene. Symptoms include growth abnormalities of the head, face and nails, along with the development of acanthosis nigricans. Treatment involves controlling blood glucose levels by using insulin and incorporating a strategically planned, controlled diet. Also, direct actions against other symptoms may be taken This syndrome usually affects children and has a prognosis of 1–2 years.
3C syndrome is a rare condition whose symptoms include heart defects, cerebellar hypoplasia, and cranial dysmorphism. It was first described in the medical literature in 1987 by Ritscher and Schinzel, for whom the disorder is sometimes named.
Antley–Bixler syndrome, is a rare, very severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.

Boomerang dysplasia is a lethal form of osteochondrodysplasia known for a characteristic congenital feature in which bones of the arms and legs are malformed into the shape of a boomerang. Death usually occurs in early infancy due to complications arising from overwhelming systemic bone malformations.

Marden–Walker syndrome (MWS) is a rare autosomal recessive congenital disorder. It is characterized by blepharophimosis, microcephaly, micrognathia, multiple joint contractures, arachnodactyly, camptodactyly, kyphoscoliosis and delayed motor development and is often associated with cystic dysplastic kidneys, dextrocardia, Dandy–Walker malformation and agenesis of corpus callosum.
Gerodermia osteodysplastica (GO), is a rare autosomal recessive connective tissue disorder included in the spectrum of cutis laxa syndromes.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency. is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.

{Short description|Medical condition}}
Cranio-lenticulo-sutural dysplasia is a neonatal/infancy disease caused by a disorder in the 14th chromosome. It is an autosomal recessive disorder, meaning that both recessive genes must be inherited from each parent in order for the disease to manifest itself. The disease causes a significant dilation of the endoplasmic reticulum in fibroblasts of the host with CLSD. Due to the distension of the endoplasmic reticulum, export of proteins from the cell is disrupted.
Malpuech facial clefting syndrome, also called Malpuech syndrome or Gypsy type facial clefting syndrome, is a rare congenital syndrome. It is characterized by facial clefting, a caudal appendage, growth deficiency, intellectual and developmental disability, and abnormalities of the renal system (kidneys) and the male genitalia. Abnormalities of the heart, and other skeletal malformations may also be present. The syndrome was initially described by Georges Malpuech and associates in 1983. It is thought to be genetically related to Juberg-Hayward syndrome. Malpuech syndrome has also been considered as part of a spectrum of congenital genetic disorders associated with similar facial, urogenital and skeletal anomalies. Termed "3MC syndrome", this proposed spectrum includes Malpuech, Michels and Mingarelli-Carnevale (OSA) syndromes. Mutations in the COLLEC11 and MASP1 genes are believed to be a cause of these syndromes. The incidence of Malpuech syndrome is unknown. The pattern of inheritance is autosomal recessive, which means a defective (mutated) gene associated with the syndrome is located on an autosome, and the syndrome occurs when two copies of this defective gene are inherited.
13q deletion syndrome is a rare genetic disease caused by the deletion of some or all of the large arm of human chromosome 13. Depending upon the size and location of the deletion on chromosome 13, the physical and mental manifestations will vary. It has the potential to cause intellectual disability and congenital malformations that affect a variety of organ systems. Because of the rarity of the disease in addition to the variations in the disease, the specific genes that cause this disease are unknown. This disease is also known as:
Kahrizi syndrome (KHRZ) is an autosomal-recessive disease that is identified by mental retardation, cataracts, coloboma, kyphosis, and coarse facial features caused by a homozygous mutation in the SRD5A3 gene.

