| Arachnodactyly | |
|---|---|
| Other names | Achromachia |
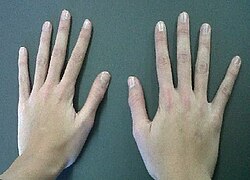 | |
| Bilateral arachnodactyly | |
| Specialty | Medical genetics |
| Complications | None |
| Usual onset | Birth |
| Duration | Life-long |
| Causes | Mutations in the fibrillin-2 gene, in chromosome 5q23, or the fibrillin-1 gene, at chromosome 15q21.1 |
Arachnodactyly ("spider fingers") is a medical condition that is characterized by fingers and toes that are abnormally long and slender, in comparison to the palm of the hand and arch of the foot. In some cases, the thumbs of an individual with the condition are pulled inwards towards the palm. This condition is present at birth.