| Osteopoikilosis | |
|---|---|
 | |
| Osteopoikilosis on an X-ray of the hands | |
| Specialty | Medical genetics |
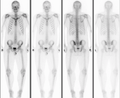
Osteopoikilosis is a benign, autosomal dominant, sclerosing (hardening) dysplasia of bone characterized by the presence of numerous bone islands in the skeleton. [1]