
Spondyloperipheral dysplasia is an autosomal dominant disorder of bone growth. The condition is characterized by flattened bones of the spine (platyspondyly) and unusually short fingers and toes (brachydactyly). Some affected individuals also have other skeletal abnormalities, short stature, nearsightedness (myopia), hearing loss, and mental retardation. Spondyloperipheral dysplasia is a subtype of collagenopathy, types II and XI.
Spondyloepimetaphyseal dysplasia, Strudwick type is an inherited disorder of bone growth that results in dwarfism, characteristic skeletal abnormalities, and problems with vision. The name of the condition indicates that it affects the bones of the spine (spondylo-) and two regions near the ends of bones. This type was named after the first reported patient with the disorder. Spondyloepimetaphyseal dysplasia, Strudwick type is a subtype of type II collagenopathies.

Tyrosine-protein kinase transmembrane receptor ROR2, also known as neurotrophic tyrosine kinase, receptor-related 2, is a protein that in humans is encoded by the ROR2 gene located on position 9 of the long arm of chromosome 9. This protein is responsible for aspects of bone and cartilage growth. It is involved in Robinow syndrome and autosomal dominant brachydactyly type B. ROR2 is a member of the receptor tyrosine kinase-like orphan receptor (ROR) family.
An osteochondrodysplasia, or skeletal dysplasia, is a disorder of the development of bone and cartilage. Osteochondrodysplasias are rare diseases. About 1 in 5,000 babies are born with some type of skeletal dysplasia. Nonetheless, if taken collectively, genetic skeletal dysplasias or osteochondrodysplasias comprise a recognizable group of genetically determined disorders with generalized skeletal affection. These disorders lead to disproportionate short stature and bone abnormalities, particularly in the arms, legs, and spine. Skeletal dysplasia can result in marked functional limitation and even mortality.

Multiple epiphyseal dysplasia (MED), also known as Fairbank's disease, is a rare genetic disorder that affects the growing ends of bones. Long bones normally elongate by expansion of cartilage in the growth plate near their ends. As it expands outward from the growth plate, the cartilage mineralizes and hardens to become bone (ossification). In MED, this process is defective.

Pseudoachondroplasia is an inherited disorder of bone growth. It is a genetic autosomal dominant disorder. It is generally not discovered until 2–3 years of age, since growth is normal at first. Pseudoachondroplasia is usually first detected by a drop of linear growth in contrast to peers, a waddling gait or arising lower limb deformities.

The sulfate transporter is a solute carrier family protein that in humans is encoded by the SLC26A2 gene. SLC26A2 is also called the diastrophic dysplasia sulfate transporter (DTDST), and was first described by Hästbacka et al. in 1994. A defect in sulfate activation described by Superti-Furga in achondrogenesis type 1B was subsequently also found to be caused by genetic variants in the sulfate transporter gene. This sulfate (SO42−) transporter also accepts chloride, hydroxyl ions (OH−), and oxalate as substrates. SLC26A2 is expressed at high levels in developing and mature cartilage, as well as being expressed in lung, placenta, colon, kidney, pancreas and testis.

Matrilin-3 is a protein that in humans is encoded by the MATN3 gene. It is linked to the development of many types of cartilage, and part of the Matrilin family, which includes Matrilin-1, Matrilin-2, Matrilin-3, and Matrilin-4, a family of filamentous-forming adapter oligomeric extracellular proteins that are linked to the formation of cartilage and bone, as well as maintaining homeostasis after development. It is considered an extracellular matrix protein that functions as an adapter protein where the Matrilin-3 subunit can form both homo-tetramers and hetero-oligomers with subunits from Matrilin-1 which is the cartilage matrix protein. This restricted tissue has been strongly expressed in growing skeletal tissue as well as cartilage and bone.

Oculodentodigital syndrome is an extremely rare genetic condition that typically results in small eyes, underdeveloped teeth, and syndactyly and malformation of the fourth and fifth fingers. It is considered a kind of ectodermal dysplasia.

Keutel syndrome (KS) is a rare autosomal recessive genetic disorder characterized by abnormal diffuse cartilage calcification, hypoplasia of the mid-face, peripheral pulmonary stenosis, hearing loss, short distal phalanges (tips) of the fingers and mild mental retardation. Individuals with KS often present with peripheral pulmonary stenosis, brachytelephalangism, sloping forehead, midface hypoplasia, and receding chin. It is associated with abnormalities in the gene coding for matrix gla protein, MGP. Being an autosomal recessive disorder, it may be inherited from two unaffected, abnormal MGP-carrying parents. Thus, people who inherit two affected MGP alleles will likely inherit KS.
Boomerang dysplasia is a lethal form of osteochondrodysplasia known for a characteristic congenital feature in which bones of the arms and legs are malformed into the shape of a boomerang. Death usually occurs in early infancy due to complications arising from overwhelming systemic bone malformations.

Frontonasal dysplasia (FND) is a congenital malformation of the midface. For the diagnosis of FND, a patient should present at least two of the following characteristics: hypertelorism, a wide nasal root, vertical midline cleft of the nose and/or upper lip, cleft of the wings of the nose, malformed nasal tip, encephalocele or V-shaped hair pattern on the forehead. The cause of FND remains unknown. FND seems to be sporadic (random) and multiple environmental factors are suggested as possible causes for the syndrome. However, in some families multiple cases of FND were reported, which suggests a genetic cause of FND.
EEM syndrome is an autosomal recessive congenital malformation disorder affecting tissues associated with the ectoderm, and also the hands, feet and eyes.
Gerodermia osteodysplastica (GO) is a rare autosomal recessive connective tissue disorder included in the spectrum of cutis laxa syndromes.

Hydrops-ectopic calcification-moth-eaten skeletal dysplasia is a defect in cholesterol biosynthesis. Greenberg characterized the condition in 1988.
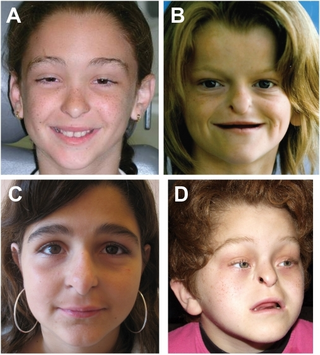
Johanson–Blizzard syndrome (JBS) is a rare, sometimes fatal autosomal recessive multisystem congenital disorder featuring abnormal development of the pancreas, nose and scalp, with intellectual disability, hearing loss and growth failure. It is sometimes described as a form of ectodermal dysplasia.

Fryns syndrome is an autosomal recessive multiple congenital anomaly syndrome that is usually lethal in the neonatal period. Fryns (1987) reviewed the syndrome.
Donnai–Barrow syndrome is a genetic disorder first described by Dian Donnai and Margaret Barrow in 1993. It is associated with LRP2. It is an inherited (genetic) disorder that affects many parts of the body.
Opsismodysplasia is a type of skeletal dysplasia first described by Zonana and associates in 1977, and designated under its current name by Maroteaux (1984). Derived from the Greek opsismos ("late"), the name "opsismodysplasia" describes a delay in bone maturation. In addition to this delay, the disorder is characterized by micromelia, particularly of the hands and feet, delay of ossification, platyspondyly, irregular metaphyses, an array of facial aberrations and respiratory distress related to chronic infection. Opsismodysplasia is congenital, being apparent at birth. It has a variable mortality, with some affected individuals living to adulthood. The disorder is rare, with an incidence of less than 1 per 1,000,000 worldwide. It is inherited in an autosomal recessive pattern, which means the defective (mutated) gene that causes the disorder is located on an autosome, and the disorder occurs when two copies of this defective gene are inherited. No specific gene has been found to be associated with the disorder. It is similar to spondylometaphyseal dysplasia, Sedaghatian type.
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although its phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.
