Ollier disease is a rare sporadic nonhereditary skeletal disorder in which typically benign cartilaginous tumors (enchondromas) develop near the growth plate cartilage. This is caused by cartilage rests that grow and reside within the metaphysis or diaphysis and eventually mineralize over time to form multiple enchondromas.[1] Key signs of the disorder include asymmetry and shortening of the limb as well as an increased thickness of the bone margin. These symptoms are typically first visible during early childhood with the mean age of diagnosis being 13 years of age.[2] Many patients with Ollier disease are prone to develop other malignancies including bone sarcomas that necessitate treatment and the removal of malignant bone neoplasm. Cases in patients with Ollier disease has shown a link to IDH1, IDH2, and PTH1R gene mutations. Currently, there are no forms of treatment for the underlying condition of Ollier disease but complications such as fractures, deformities, malignancies that arise from it can be treated through surgical procedures. The prevalence of this condition is estimated at around 1 in 100,000.[3] It is unclear whether the men or women are more affected by this disorder due to conflicting case studies.
The disease consists of the growth multiple enchondromas which usually develop in early childhood. The growth of these enchondromas usually stops after skeletal maturation.[4] The affected extremity is shortened (asymmetric dwarfism) and sometimes bowed due to epiphyseal fusion anomalies. Bone lesions generally present as cellular during childhood and become more solitary over time. People with Ollier disease are prone to breaking bones (fractures) and normally have swollen, aching limbs. However, many cases of solitary enchondromata go unnoticed due to lack of symptoms. Enchondromas are commonly found in the phalanges, metacarpal, and metatarsal bones in patients of Ollier disease due to the affinity of enchondromas to long tubular bones such as the femur and humerus. A unilateral distribution of bone lesions is usually observed but bilateral distributions or a singular extremity can occur as well. Approximately a third of the cases show some form of physical deformities of bowing or abnormal limb lengthening.[citation needed]
Associated conditions
Ollier disease carries a higher risk of malignancies such as central nervous system (CNS), ovarian, and adenocarcinoma.[5] Cranial gliomas have been linked with this disorder at an increased rate and at an earlier diagnosis age.[6][7][8] A majority of glioma cases contain IDH gene mutations thus explaining the link between the two conditions. Juvenile granulosa cell tumour has also been associated with the disease.[9] One case study indicates that this is due a mesodermal dysplasia in the long bones resulting in ovarian cancer.[10]
The incidence of a secondary chondrosarcoma in Ollier disease is most commonly approximated at 25–30% with some projections even as high as 50%.[11][12][13] Chondrosarcomas are typically developed during young adulthood and mostly form as a unifocal distribution in Ollier disease patients.[3] The most common locations of tumors are in the pelvis and shoulder girdle.[14] While chondrosarcoma is the most common form of a secondary malignant bone neoplasm found in cases of Ollier disease, other forms such as chordomas and osteosarcomas can occur. If left untreated, these malignant transformations may lead to fatal outcomes.[citation needed]
A related and even rarer disorder named Maffucci syndrome is a very similar condition that is characterized by the presence of multiple enchondromas with hemangiomas and occasionally lymphangiomas usually near the hands and feet but not limited to the skull, ribs, and spinal bones.[2] This disorder is also sporadic and nonhereditary and usually detected during childhood despite being a congenital condition. Maffucci syndrome has also similarly been linked to IDH1 and IDH2 mutations specifically the IDH1 R132C hotspot mutation.[15] This hotspot mutation is presumed to be responsible for the spindle cell hemangiomas and enchondromas in cases of Maffucci syndrome. Sanger sequencing analysis concluded that exon 4 is the primary location of mutations in IDH1 and IDH2 genes are specifically responsible for hemangiomas. Maffucci syndrome carries a significantly higher risk of malignant transformations like chondrosarcomas but also much more aggressive tumors such as acute lymphocytic leukemia and gastrointestinal and ovarian malignancies.[1][16]
Cause
For many years, most research has been inconclusive regarding the cause of the disease.[1][17]
Recent studies have shown that most cases of Ollier disease are believed to have been caused by isocitrate dehydrogenases IDH1 and IDH2 mutations.[18] In one study, 35 of 43 (81%) patients with Ollier disease had either a IDH1 or IDH2 mutation.[19] Another study suggests that R132C IDH1 mutations which are particularly dominant at exon 4 of IDH genes are linked to the growth of vascular lesions.[15] Isocitrate deyhydrogenases IDH1 and IDH2 are catalysts responsible for the conversion of isocitrate to 2-oxoglutarate. The isocitrate deyhydrogenases IDH1 and IDH2 mutations disrupt this process resulting in unregulated production of α-ketoglutarate and a reduction in chondrocyte proliferation. In many cases of cartilaginous tumors, IDH1 and IDH2 point mutations were found thus explaining why Ollier disease is associated with many different associated conditions. Based on these case studies, most evidence suggests that the abnormal lining of lesions found in Ollier disease would suggest that the condition is caused by a post-zygotic somatic mutation thus resulting in a mosaic genetic disorder.[20]
Approximately 8–10% of cases of patients with Ollier disease have been linked to PTH1R mutations.[17][19] A particular case study the mutant heterozygous PTHR1 (R150C) receptor was observed in two unrelated patients with Ollier disease.[21][22] This PTHR1 (R150C) mutant causes a reduction in chondrocyte differentiation by triggering the PTHrP-dependent pathway and decreasing PTHLH receptor function by approximately 30% creating enchondromas. One of these patients with the PTHR1 (R150C) mutant was found to have inherited the mutation from his father. This provides credence to the theory that multiple genetic mutations are needed to occur in order for Ollier disease to manifest.
An alternative theory suggests that since there have been cases of multiple family members with enchondromatosis, the disorder may be passed on through autosomal dominant inheritance.[1]
Diagnosis
Clinical and radiological evaluations are conducted in order to detect the presence of bone neoplasms or lesions typically found in Ollier disease. Histological evaluations are mainly used to examine or detect malignancies.
Abnormal bone growth such as shortening or thickening and deformity may be observed in patients of Ollier disease. These bone lesions are visible at birth using radiography but are usually not screened or examined for until clinical manifestations present during early childhood. However, some patients may exhibit no signs of any symptoms.[1] One study found thirteen to be the mean age of diagnosis in patients with Ollier disease. In an X-ray, there would normally be the presence of several homogeneous lesions of an oval or elongated shape with bone edges that are slightly thickened.[3] With age, these lesions may calcify and appear as diffusely minute spots or stippled. Fan-like septations or streaks would be indicative of the presence of several enchondromas. Early detection and consistent and repeated monitoring is important in order to prevent and treat any potential bone neoplasms.[citation needed]
Magnetic resonance imaging (MRI), ultrasound, and scintigraphy are generally not practical for diagnostic purposes. X-rays are not as effective in the monitoring or evaluation of enchondromas due to frequent localized changes also sometimes due to the large number of enchondromas. MRI can sometimes however be used to monitor and evaluate symptomatic lesions in the case of potential malignant transformations.
Similar disorders such as Maffucci syndrome and hereditary multiple exostoses (HME) require differentiation during diagnosis. Maffucci syndrome can be distinguished clinically by the presence of hemangiomas and lymphangiomas and genetically through R132C IDH1 hotspot mutations.[15] HME features osteochondromas which are near the surface the bone whereas enchondromas featured in Ollier disease and Maffucci are more towards the center of the bone.[3] Also, neural compressions are more commonly found in HME than in Ollier disease.
Treatment
The condition of Ollier disease cannot be treated for but the complications that arise such as fractures, growth defects, and tumors can be surgically treated. These are typically done to treat and remove any extraneous bone tissue while preserving the function of limb if possible.[11]
Fractures have been treated using a variety of methods such as bone grafting, internal fixation, corticoplasty, Ilizarov technique, elastic stable intramedullary nailing system (ESIN), and flexible intramedullary nailing (FIN).
Corticoplasty has been shown to have success in treating hand lesions and deformities while retaining normal function.[23][24] The surgery utilizes the removal of tissue (curettage) and bone reconstruction in order to remove enchondromas and improve cosmetic appearance. Many cases of corticoplasty have been shown improvement in appearances while maintaining function. Recurrence of enchondromas was observed in some cases. In cases of Ollier disease, early surgical treatment of enchondromas in the hand is recommended.
The Ilizarov technique is a form of noninvasive treatment that can sometimes be used to reshape and correct deformities and misaligned limb bones.[25][26] It uses the process of external fixation through the scaffolding of pins or wires into the bone in an attempt to transform enchondromata into normal bone tissue. This method of treatment is very safe but is also very strenuous as it a long-term procedure. The ring fixators used in the Ilizarov technique have been shown to be very versatile for realigning and lengthening limb bones while also managing soft tissue tension.[27] Complications from this procedure may include a recurrence in limb lengthening complications such as premature healing requiring osteoclasis and a return of angular deformities. Reduced latency periods followed by faster distraction times are encouraged in order to prevent premature healing. The Ilizarov technique has shown to have positive outcomes for many cases of Ollier disease with some patients even experiencing full correction of deformity and length in best cases scenarios.
Elastic stable intramedullary nailing system (ESIN) and flexible intramedullary nailing (FIN) are more recent surgical procedures that utilize internal fixation that has been shown to reduce the Healing Index and minimize complications.[28][29] Similarly to the Ilizarov technique, the procedure is intended to correct deformities and elongate limb bones. This form of treatment is not suitable for the elongation of bones with a small shaft diameter or sections containing open growth zones. Patients with severe forms of Ollier disease also are not suited for surgery because of an increased risk of complications due to bone frame instability. Both the elastic stable intramedullary nailing system (ESIN) and flexible intramedullary nailing (FIN) use two bent elastic nails in order to allow for greater realignment and stability. This technique used in conjunction with a circular external fixator has been shown to significantly reduce Healing Index values in both monosegmental and polysegmental lengthening. Another advantage is that no fractures or deformities were found in later follow ups of 2 to 5 years.
Elastic stable intramedullary nailing system (ESIN) requires a longer duration of external fixation for most procedures compared to flexible intramedullary nailing (FIN).[citation needed]
Malignant transformations of any or multiple enchondromas are common in patients with Ollier disease and typically emerge in young adults often requiring surgery. The average age of patients of Ollier disease for their first surgery to treat their chondrosarcoma is thirty-three. Some examples of surgical procedures performed to treat secondary malignant bone neoplasms from Ollier disease include amputation, wide-local excisions, hemipelvectomy, and arthroplasty.[30] Cobalt and chemotherapy are typically not primary methods of surgery as chondrosarcomas generally do not have enough blood supply to make it an effective form of treatment.[31] These are done to treat and remove any extraneous bone tissue while preserving the function of limb if possible.[11]
Epidemiology
One out of every 100,000 people is estimated to have Ollier disease. However, this estimate may be low due to under reporting of cases from asymptomatic or mild conditions.[17] There are many contradicting studies that report different prevalence rates between genders as one study indicates that it affects both sexes equally while other studies purporting that it affects men or women more frequently.[17][8][20] The average age of diagnosis is 13 years. Ollier disease is not normally diagnosed until early childhood due to lack of visible symptoms present at birth despite lesions being present using radiography. Although most research suggests that Ollier disease is spontaneous and nonhereditary, there are some cases where it appears among family members.
Eponyms
The disorder is named after French surgeonLouis Léopold Ollier.[32] Late in the 19th century, Ollier was one of the first to distinguish between enchondromatosis and this condition by highlighting the pattern of abnormal and asymmetrical enchondromas distributions in Ollier disease.[3]
Gallery
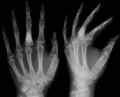
X-ray showing calcified enchondromas localized in finger a 37-year-old patient affected with Ollier disease
X-ray showing enchondromas localized in the humerus of a 37-year-old patient affected with Ollier disease
X-ray showing enchondromas localized in the lower part of the radius of a 37-year-old patient affected with Ollier disease
MRI showing enchondromas localized in the lower part of the radius of a 37-year-old patient affected with Ollier disease
MRI showing enchondromas localized in the lower part of the radius of a 37-year-old patient affected with Ollier disease.
Enchondromas localized in the upper part of the humerus of the same patient
1 2 3 4 5 Muthusamy, Saravanaraja; Conway, Sheila A.; Temple, H. Thomas (July 2014). "Five Polyostotic Conditions That General Orthopedic Surgeons Should Recognize (or Should Not Miss)". Orthopedic Clinics of North America. 45 (3): 417–429. doi:10.1016/j.ocl.2014.04.004. ISSN0030-5898. PMID24975767.
↑ Turek's orthopaedics principles and their application (6thed.). Philadelphia: Lippincott Williams & Wilkins. 2005. p.438. ISBN9780781742986.
↑ Beckingsale, Thomas B.; Shaw, Colin (June 2015). "(v) Epidemiology of bone & soft tissue sarcomas". Orthopaedics and Trauma. 29 (3): 182–188. doi:10.1016/j.mporth.2015.03.001. ISSN1877-1327.
↑ Halal, Fahed; Azouz, E. Michel (1991-03-15). "Generalized enchondromatosis in a boy with only platyspondyly in the father". American Journal of Medical Genetics. 38 (4): 588–592. doi:10.1002/ajmg.1320380418. ISSN0148-7299. PMID2063903.
↑ Hopyan, Sevan; Gokgoz, Nalan; Poon, Raymond; Gensure, Robert C.; Yu, Chunying; Cole, William G.; Bell, Robert S.; Jüppner, Harald; Andrulis, Irene L.; Wunder, Jay S.; Alman, Benjamin A. (2002-02-19). "A mutant PTH/PTHrP type I receptor in enchondromatosis". Nature Genetics. 30 (3): 306–310. doi:10.1038/ng844. ISSN1061-4036. PMID11850620. S2CID5961561.
↑ Kim, Eugene; Miyake, Junichi; Kataoka, Toshiyuki; Oka, Kunihiro; Moritomo, Hisao; Murase, Tsuyoshi (November 2012). "Corticoplasty for Improved Appearance of Hands With Ollier Disease". The Journal of Hand Surgery. 37 (11): 2294–2299. doi:10.1016/j.jhsa.2012.08.006. ISSN0363-5023. PMID23040642.
↑ Van Loon, Pieter; Lammens, Johan (March 2008). "Malformation of the humerus in a patient with Ollier disease treated with the Ilizarov technique". Journal of Shoulder and Elbow Surgery. 17 (2): e9 –e11. doi:10.1016/j.jse.2007.04.006. ISSN1058-2746. PMID18036843.
↑ Watanabe, Koji; Tsuchiya, Hiroyuki; Sakurakichi, Keisuke; Yamashiro, Teruhisa; Matsubara, Hidenori; Tomita, Katsuro (September 2007). "Treatment of lower limb deformities and limb-length discrepancies with the external fixator in Ollier's disease". Journal of Orthopaedic Science. 12 (5): 471–475. doi:10.1007/s00776-007-1163-9. ISSN0949-2658. PMID17909933. S2CID25701102.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.