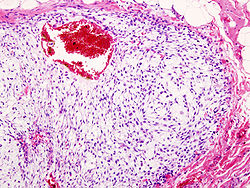
Chondrosarcoma is a bone sarcoma, a primary cancer composed of cells derived from transformed cells that produce cartilage.[1] A chondrosarcoma is a member of a category of tumors of bone and soft tissue known as sarcomas. About 30% of bone sarcomas are chondrosarcomas.[2] It is resistant to chemotherapy and radiotherapy with mesenchymal suptype being a notable exception.[3] Unlike other primary bone sarcomas that mainly affect children and adolescents, a chondrosarcoma can present at any age. It more often affects the axial skeleton than the appendicular skeleton.[4]
MRI of a left-pelvis chondrosarcoma in a 26-year-old maleMetastatic chondrosarcoma at the lower lip
Imaging studies – including radiographs ("x-rays"), computerized tomography (CT), and magnetic resonance imaging (MRI) – are often used to make a presumptive diagnosis of chondrosarcoma.[10] However, a definitive diagnosis depends on the identification of malignant cancer cells producing cartilage in a biopsy specimen that has been examined by a pathologist. In a few cases, usually of highly anaplastic tumors, immunohistochemistry (IHC) is required.[11]
There are no blood tests currently available to enable an oncologist to render a diagnosis of chondrosarcoma. The most characteristic imaging findings are usually obtained with CT.[12]
Nearly all chondrosarcoma patients appear to be in good health. Often, patients are not aware of the growing tumor until there is a noticeable lump or pain. Earlier diagnosis is generally accidental when a patient undergoes testing for another problem and physicians discover cancer. Occasionally the first symptom will be a broken bone at the cancerous site. Any broken bone that occurs from mild trauma warrants further investigation, although there are many conditions that can lead to weak bones, and this form of cancer is not a common cause of such breaks.[13]
Treatment
Treatment depends on the location of the disease and the aggressiveness of the tumors.[14]
Surgery is the main form of treatment for chondrosarcoma. Musculoskeletal tumor specialists or orthopedic oncologists are usually chosen to treat chondrosarcoma, unless it is located in the skull, spine, or chest cavity, in which case, a neurosurgeon or thoracic surgeon experienced with sarcomas is chosen. Often, a limb-sparing operation can be performed, but in some cases amputation is unavoidable. Amputation of the arm, leg, jaw, or half of the pelvis (called a hemipelvectomy) may be necessary in some cases.[15]
There are two kinds of hemipelvectomy – internal and external.
External hemipelvectomy – is removal of that half of the pelvis with the amputation of the leg. It is also called hindquarter amputation.
Internal hemipelvectomy – is removal of that half of the pelvis, but the leg is left intact.
Amputation at the hip is called hip disarticulation and amputees who have had this amputation are also called hip disartics.
Chemotherapy or traditional radiotherapy are not very effective for most chondrosarcomas, although proton therapy is showing promise with local tumor control at over 80%.[16] The only potentially chemosensitive chondrosarcoma subtypes are dedifferentiated and mesenchymal. [17][18]
Complete surgical ablation is the most effective treatment, but sometimes this is difficult. Proton therapy radiation can be useful in awkward locations to make surgery more effective.
Recent studies have shown that induction of apoptosis in high-grade chondrosarcoma, both directly and by enhancement of response to chemotherapy and radiation, is a valid therapeutic strategy.[19]
Prognosis
Prognosis depends on how early the cancer is discovered and treated. For the least aggressive grade, about 90% of patients survive more than five years after diagnosis. People usually have a good survival rate at the low-grade volume of cancer.[4] For the most aggressive grade, only 10% of patients will survive one year. Tumors may recur in the future. Follow up scans are extremely important for chondrosarcoma to make sure there has been no recurrence or metastasis, which usually occurs in the lungs.[20]
Chondrosarcoma in Art
The memoir-in-verse No Obvious Distress is about poet Amanda Quaid's journey with mesenchymal chondrosarcoma.[21][22]
12Pathology and genetics of tumours of soft tissue and bone. Fletcher, Christopher D. M., Unni, K. Krishnan, 1941–, Mertens, Fredrik., World Health Organization., International Agency for Research on Cancer. Lyon: IARC Press. 2002. ISBN978-92-832-2413-6. OCLC51001831.{{cite book}}: CS1 maint: others (link)
↑Amary, M. Fernanda; Bacsi, Krisztian; Maggiani, Francesca; Damato, Stephen; Halai, Dina; Berisha, Fitim; Pollock, Robin; O'Donnell, Paul; Grigoriadis, Anita (Jul 2011). "IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours". The Journal of Pathology. 224 (3): 334–343. doi:10.1002/path.2913. hdl:1887/108488. ISSN1096-9896. PMID21598255. S2CID24201208.
↑Biermann, J. Sybil; Chow, Warren; Reed, Damon R.; Lucas, David; Adkins, Douglas R.; Agulnik, Mark; Benjamin, Robert S.; Brigman, Brian; Budd, G. Thomas; Curry, William T.; Didwania, Aarati; Fabbri, Nicola; Hornicek, Francis J.; Kuechle, Joseph B.; Lindskog, Dieter (February 2017). "NCCN Guidelines Insights: Bone Cancer, Version 2.2017". Journal of the National Comprehensive Cancer Network. 15 (2): 155–167. doi:10.6004/jnccn.2017.0017. ISSN1540-1405. PMID28188186.
↑Jamil N, Howie S, Salter DM. Therapeutic molecular targets in human chondrosarcoma .Int J Exp Pathol 2010; 91:387–93
↑Limaiem F, Davis D, Sticco K (2021). "Chondrosarcoma". National Center for Biotechnology Information, U.S. National Library of Medicine. PMID30844159. Retrieved 8 July 2021.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.