
Chromosome 15q partial deletion is a rare human genetic disorder, caused by a chromosomal aberration in which the long ("q") arm of one copy of chromosome 15 is deleted, or partially deleted. Like other chromosomal disorders, this increases the risk of birth defects, developmental delay and learning difficulties, however, the problems that can develop depend very much on what genetic material is missing. If the mother's copy of the chromosomal region 15q11-13 is deleted, Angelman syndrome (AS) can result. The sister syndrome Prader-Willi syndrome (PWS) can result if the father's copy of the chromosomal region 15q11-13 is deleted. The smallest observed region that can result in these syndromes when deleted is therefore called the PWS/AS critical region. In addition to deletions, uniparental disomy of chromosome 15 also gives rise to the same genetic disorders, indicating that genomic imprinting must occur in this region.

Myalgia is the medical term for muscle pain. Myalgia is a symptom of many diseases. The most common cause of acute myalgia is the overuse of a muscle or group of muscles; another likely cause is viral infection, especially when there has been no trauma.

Menkes disease (MNK), also known as Menkes syndrome, is an X-linked recessive disorder caused by mutations in genes coding for the copper-transport protein ATP7A, leading to copper deficiency. Characteristic findings include kinky hair, growth failure, and nervous system deterioration. Like all X-linked recessive conditions, Menkes disease is more common in males than in females. The disorder was first described by John Hans Menkes in 1962.

Aase syndrome or Aase–Smith syndrome is a rare inherited disorder characterized by anemia with some joint and skeletal deformities. Aase syndrome is thought to be an autosomal dominant inherited disorder. The genetic basis of the disease is not known. The anemia is caused by underdevelopment of the bone marrow, which is where blood cells are formed.

Angiomas are benign tumors derived from cells of the vascular or lymphatic vessel walls (endothelium) or derived from cells of the tissues surrounding these vessels.

Enchondroma is a type of benign bone tumor belonging to the group of cartilage tumors. There may be no symptoms, or it may present typically in the short tubular bones of the hands with a swelling, pain or pathological fracture.
Hypochondrogenesis is a severe genetic disorder causing malformations of bone growth. The condition is characterized by a short body and limbs and abnormal bone formation in the spine and pelvis.
Metachondromatosis is an autosomal dominant, incompletely penetrant genetic disease affecting the growth of bones, leading to exostoses primarily in the hands and feet as well as enchondromas of long bone metaphyses and iliac crests. This syndrome affects mainly tubular bones, though it can also involve the vertebrae, small joints, and flat bones. The disease is thought to affect exon 4 of the PTPN11 gene. Metachondromatosis is believed to be caused by an 11 base pair deletion resulting in a frameshift and nonsense mutation. The disease was discovered and named in 1971 by Pierre Maroteaux, a French physician, when he observed two families with skeletal radiologic features with exostoses and Ollier disease. The observation of one family with five affected people led to the identification of the disease as autosomal dominant. There have been less than 40 cases of the disease reported to date.
Jackson–Weiss syndrome (JWS) is a genetic disorder characterized by foot abnormalities and the premature fusion of certain bones of the skull (craniosynostosis), which prevents further growth of the skull and affects the shape of the head and face. This genetic disorder can also sometimes cause intellectual disability and crossed eyes. It was characterized in 1976.
Bannayan–Riley–Ruvalcaba syndrome (BRRS) is a rare overgrowth syndrome and hamartomatous disorder with occurrence of multiple subcutaneous lipomas, macrocephaly and hemangiomas. The disease is inherited in an autosomal dominant manner. The disease belongs to a family of hamartomatous polyposis syndromes, which also includes Peutz–Jeghers syndrome, juvenile polyposis and Cowden syndrome. Mutation of the PTEN gene underlies this syndrome, as well as Cowden syndrome, Proteus syndrome, and Proteus-like syndrome, these four syndromes are referred to as PTEN Hamartoma-Tumor Syndromes.
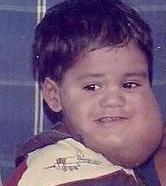
A cystic hygroma is an abnormal growth that usually appears on a baby's neck or head. It consists of one or more cysts and tends to grow larger over time. The disorder usually develops while the fetus is still in the uterus, but can also appear after birth.
Osteochondrodysplasia is a general term for a disorder of the development (dysplasia) of bone ("osteo") and cartilage ("chondro"). Osteochondrodysplasias are rare diseases. About 1 in 5,000 babies are born with some type of skeletal dysplasia. Nonetheless, if taken collectively, genetic skeletal dysplasias or osteochondrodysplasias comprise a recognizable group of genetically determined disorders with generalized skeletal affection. Osteochondrodysplasias can result in marked functional limitation and even mortality.

Ollier disease is a rare sporadic nonhereditary skeletal disorder in which typically benign cartilaginous tumors (enchondromas) develop near the growth plate cartilage. This is caused by cartilage rests that grow and reside within the metaphysis or diaphysis and eventually mineralize over time to form multiple enchondromas. Key signs of the disorder include asymmetry and shortening of the limb as well as an increased thickness of the bone margin. These symptoms are typically first visible during early childhood with the mean age of diagnosis being 13 years of age. Many patients with Ollier disease are prone to develop other malignancies including bone sarcomas that necessitate treatment and the removal of malignant bone neoplasm. Cases in patients with Ollier disease has shown a link to IDH1, IDH2, and PTH1R gene mutations. Currently, there are no forms of treatment for the underlying condition of Ollier disease but complications such as fractures, deformities, malignancies that arise from it can be treated through surgical procedures. The prevalence of this condition is estimated at around 1 in 100,000. It is unclear whether the men or women are more affected by this disorder due to conflicting case studies.

Angelo Maria Maffucci was an Italian pathologist of the nineteenth century. His most important scientific contribution is related to the description of the disease known as Maffucci's Syndrome. Maffucci was a pioneer in the field of embryonal infective pathology. His settlement in Pisa, as the chairman of Pathology, represents a very significant moment for the Pisan academic environment and for the University of Pisa.
Schinzel–Giedion syndrome (SGS) is a congenital neurodegenerative terminal syndrome. It was first described in 1978 by Albert Schinzel (1944–) and Andreas Giedion (1925–) as a syndrome with severe midface retraction, skull anomalies, renal anomalies (hydronephrosis) and other anomalies. Babies born with Schinzel–Giedion syndrome have severe mental retardation, growth retardation and global developmental delay.

In medical contexts, a facies is a distinctive facial expression or appearance associated with a specific medical condition. The term comes from Latin for "face". As a fifth declension noun, facies can be both singular and plural.
Laryngo-onycho-cutaneous syndrome is a rare epithelial disorder inherited in an autosomal recessive fashion. It is characterized by abnormalities in the larynx, nails, and skin ("cutaneous"). The disorder is only found in Punjabi Muslims and only a few cases have been reported.

Osteoporosis-pseudoglioma syndrome or OPGG is a rare genetic condition which is characterized by early-onset blindness and severe osteoporosis alongside seemingly random bone fractures.
Spastic paraplegia 31 is a rare type of hereditary spastic paraplegia which is characterized by sensation anomalies of the lower extremities.
Spondyloenchondrodysplasia is the medical term for a rare spectrum of symptoms that are inherited following an autosomal recessive inheritance pattern. Skeletal anomalies are the usual symptoms of the disorder, although its phenotypical nature is highly variable among patients with the condition, including symptoms such as muscle spasticity or thrombocytopenia purpura. It is a type of immunoosseous dysplasia.
