The natural source of galantamine are certain species of daffodil and because these species are scarce and because the isolation of galanthamine from daffodil is expensive (a 1996 figure specifies 50,000 US dollar per kilogram, the yield from daffodil is 0.1–0.2% dry weight) alternative synthetic sources are under development by means of total synthesis.
Outline
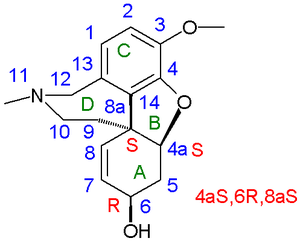
Galanthamine numbering scheme and stereocenters
In 1962 racemic galanthamine and epi-galanthamine were prepared by organic reduction of racemic narwedine by D. H. R. Barton. Narwedine is the related enone (galanthamine the allyl alcohol) obtained in an oxidative coupling. Chemical yield: 1.4%. In addition they isolated (−)-narwardine by chiral resolution from a mixture of racemic narwedine and 0.5 equivalents of (+)-galanthamine. In this way they were able to obtain (−)galanthamine again by reduction In 1976 Kametani obtained both galanthamine enantiomers by using a derivative of tartaric acid as a chiral resolving agent. In 1977 Koga obtained both enantiomers via a chiral pool synthesis starting from L-tyrosine[2][3] and in 1988 Carrol optimized the oxidative coupling route to 11% yield based on isovanillin.
In 1989 Vlahov exploited asymmetric reduction by biocatalysis in the synthesis of several galanthamine precursors. and in 1994 Shieh & Carlson [4] obtained (−)-galanthamine by spontaneous resolution of its narwedine precursor. Racemic narwedine was treated with 0.01 equivalent of (+)-galanthamine resulting in a 76% yield. Narwedine is a racemic conglomerate allowing the isolation of the S,S enantiomer from the R,R enantiomer by simple crystallization. What made the process unique is that both enantiomers are in dynamic chemical equilibrium with each other through a common phenol in a Michael reaction-like reaction brought about by triethylamine.
Resolution of Narwedine
Resolution of Narwedine
In 1999 Jordis performed (−)-galanthamine synthesis on a multikilogram scale based on Carrol chemistry and Shieh/Carlson chiral resolution. This would become the basis for current industrial production by Sanochemia (AT). In 2000 Fels proposed an intramolecularHeck reaction for the construction of the galanthamine backbone and in the same year Trost & Toste obtained (−)-galanthamine in an asymmetric synthesis involving asymmetric allylic alkylation and an intramolecular Heck reaction. Improved methods were published in 2002 and 2005 (see below) In 2004 Node obtained (−)-galanthamine via a remote asymmetric induction method with starting chiral compound D-phenylalanine.[5] Brown prepared (−)-galanthamine in 2007 starting from isovanillin.[6] Isovanillin was also used by Magnus (2009) [7] D-glucose was used by Chida (2010).[8]
Syntheses of racemic galanthamine have been reported by Wang in 2006 [9] and by Saito in 2008.[10]
Sanochemia industrial production
The method outlined by Jordis in 1999 forms the basis for industrial galanthamine production.[11]
Enantiopure (−)-narwedine is obtained via the dynamic chiral resolution method pioneered by Shieh/Carlson and in the final step the ketone is reduced to the alcohol with L-selectride.
reduction of (−)-narwedine to (−)-galanthamine as the bromide
reduction of (−)-narwedine to (−)-galanthamine as the bromide
This final step is enantioselective producing the desired S,S,R compound because the approach of H− is restricted to the Si face as the Re face is shielded by the DB ring system. Formation of the S,S,S epimer is also avoided by keeping the reaction temperature below −15°C.
↑ Synthesis and Pharmacology of Galantamine José Marco-Contelles, Maria do Carmo Carreiras, Carolina Rodríguez, Mercedes Villarroya, and Antonio G. García Chem. Rev.; 2006; 106(1) pp 116–133; (Review) doi:10.1021/cr040415t
↑ Koga, Kenji; Tomioka, Kiyoshi; Shimizu, Kimihiro; Yamada, Shun-Ichi (1977). "Approaches to the Biogenetic-type Asymmetric Synthesis of Some Amaryllidaceae Alkaloids". Heterocycles. 6 (9): 1752. doi:10.3987/R-1977-09-1752 (inactive 2024-02-17).{{cite journal}}: CS1 maint: DOI inactive as of February 2024 (link)
↑ Koga, Kenji; Shimizu, Kimihiro; Tomioka, Kiyoshi; Yamada, Shun-Ichi (1977). "A Biogenetic-type Asymmetric Synthesis of Optically Active Amaryllidaceae Alkaloids: (+)- and (−)-Galanthamine from L-Tyrosine". Heterocycles. 8: 277. doi:10.3987/S(S)-1977-01-0277 (inactive 2024-02-17).{{cite journal}}: CS1 maint: DOI inactive as of February 2024 (link)
↑ Asymmetric Transformation of Either Enantiomer of Narwedine via Total Spontaneous Resolution Process, a Concise Solution to the Synthesis of (−)-Galanthamine Wen-Chung Shieh and John A. Carlson J. Org. Chem.; 1994; 59(18) pp 5463–5465; doi:10.1021/jo00097a060
↑ Kodama, Sumiaki; Hamashima, Yoshio; Nishide, Kiyoharu; Node, Manabu (2004). "Total Synthesis of (−)-Galanthamine by Remote Asymmetric Induction". Angewandte Chemie International Edition. 43 (20): 2659–2661. doi:10.1002/anie.200353636. PMID18629982.
1 2 Satcharoen, Vachiraporn; McLean, Neville J.; Kemp, Stephen C.; Camp, Nicholas P.; Brown, Richard C. D. (2007). "Stereocontrolled Synthesis of (−)-Galanthamine". Organic Letters. 9 (10): 1867–1869. doi:10.1021/ol070255i. PMID17429978.
↑ Magnus, Philip; Sane, Neeraj; Fauber, Benjamin P.; Lynch, Vince (2009). "Concise Syntheses of (−)-Galanthamine and (±)-Codeine via Intramolecular Alkylation of a Phenol Derivative". Journal of the American Chemical Society. 131 (44): 16045–16047. doi:10.1021/ja9085534. PMID19835379.
↑ Ishikawa, Teruhiko; Kudo, Kazuhiro; Kuroyabu, Ken; Uchida, Satoshi; Kudoh, Takayuki; Saito, Seiki (2008). "Domino Double Michael−Claisen Cyclizations: A Powerful General Tool for Introducing Quaternary Stereocenters at C(4) of Cyclohexane-1,3-diones and Total Synthesis of Diverse Families of Sterically Congested Alkaloids". The Journal of Organic Chemistry. 73 (19): 7498–7508. doi:10.1021/jo801316s. PMID18781800.
↑ Development of a Pilot Scale Process for the Anti-Alzheimer Drug (−)-Galanthamine Using Large-Scale Phenolic Oxidative Coupling and Crystallisation-Induced Chiral Conversion Bernhard Küenburg, Laszlo Czollner, Johannes Fröhlich, and Ulrich Jordis Org. Process Res. Dev.; 1999; 3(6) pp 425–431; (Article) doi:10.1021/op990019q
In chemistry, an enantiomer – also called optical isomer, antipode, or optical antipode – is one of two stereoisomers that are nonsuperposable onto their own mirror image. Enantiomers of each other are much like one's right and left hands; without mirroring one of them, hands cannot be superposed onto each other. It is solely a relationship of chirality and the permanent three-dimensional relationships among molecules or other chemical structures: no amount of re-orientiation of a molecule as a whole or conformational change converts one chemical into its enantiomer. Chemical structures with chirality rotate plane-polarized light. A mixture of equal amounts of each enantiomer, a racemic mixture or a racemate, does not rotate light.
In organic chemistry, the ene reaction is a chemical reaction between an alkene with an allylic hydrogen and a compound containing a multiple bond, in order to form a new σ-bond with migration of the ene double bond and 1,5 hydrogen shift. The product is a substituted alkene with the double bond shifted to the allylic position.
In chemistry, stereoselectivity is the property of a chemical reaction in which a single reactant forms an unequal mixture of stereoisomers during a non-stereospecific creation of a new stereocenter or during a non-stereospecific transformation of a pre-existing one. The selectivity arises from differences in steric and electronic effects in the mechanistic pathways leading to the different products. Stereoselectivity can vary in degree but it can never be total since the activation energy difference between the two pathways is finite: both products are at least possible and merely differ in amount. However, in favorable cases, the minor stereoisomer may not be detectable by the analytic methods used.
1,1′-Bi-2-naphthol (BINOL) is an organic compound that is often used as a ligand for transition-metal catalysed asymmetric synthesis. BINOL has axial chirality and the two enantiomers can be readily separated and are stable toward racemisation. The specific rotation of the two enantiomers is 35.5° (c = 1 in THF), with the R enantiomer being the dextrorotary one. BINOL is a precursor for another chiral ligand called BINAP. The volumetric mass density of the two enantiomers is 0.62 g cm−3.
The Corey–Itsuno reduction, also known as the Corey–Bakshi–Shibata (CBS) reduction, is a chemical reaction in which a prochiral ketone is enantioselectively reduced to produce the corresponding chiral, non-racemic alcohol. The oxazaborolidine reagent which mediates the enantioselective reduction of ketones was previously developed by the laboratory of Itsuno and thus this transformation may more properly be called the Itsuno-Corey oxazaborolidine reduction.
Aflatoxin total synthesis concerns the total synthesis of a group of organic compounds called aflatoxins. These compounds occur naturally in several fungi. As with other chemical compound targets in organic chemistry, the organic synthesis of aflatoxins serves various purposes. Traditionally it served to prove the structure of a complex biocompound in addition to evidence obtained from spectroscopy. It also demonstrates new concepts in organic chemistry and opens the way to molecular derivatives not found in nature. And for practical purposes, a synthetic biocompound is a commercial alternative to isolating the compound from natural resources. Aflatoxins in particular add another dimension because it is suspected that they have been mass-produced in the past from biological sources as part of a biological weapons program.
The Carroll rearrangement is a rearrangement reaction in organic chemistry and involves the transformation of a β-keto allyl ester into a α-allyl-β-ketocarboxylic acid. This organic reaction is accompanied by decarboxylation and the final product is a γ,δ-allylketone. The Carroll rearrangement is an adaptation of the Claisen rearrangement and effectively a decarboxylative allylation.
In stereochemistry, a chiral auxiliary is a stereogenic group or unit that is temporarily incorporated into an organic compound in order to control the stereochemical outcome of the synthesis. The chirality present in the auxiliary can bias the stereoselectivity of one or more subsequent reactions. The auxiliary can then be typically recovered for future use.
In chemistry, transfer hydrogenation is a chemical reaction involving the addition of hydrogen to a compound from a source other than molecular H2. It is applied in laboratory and industrial organic synthesis to saturate organic compounds and reduce ketones to alcohols, and imines to amines. It avoids the need for high-pressure molecular H2 used in conventional hydrogenation. Transfer hydrogenation usually occurs at mild temperature and pressure conditions using organic or organometallic catalysts, many of which are chiral, allowing efficient asymmetric synthesis. It uses hydrogen donor compounds such as formic acid, isopropanol or dihydroanthracene, dehydrogenating them to CO2, acetone, or anthracene respectively. Often, the donor molecules also function as solvents for the reaction. A large scale application of transfer hydrogenation is coal liquefaction using "donor solvents" such as tetralin.
The Meerwein–Ponndorf–Verley (MPV) reduction in organic chemistry is the reduction of ketones and aldehydes to their corresponding alcohols utilizing aluminium alkoxide catalysis in the presence of a sacrificial alcohol. The advantages of the MPV reduction lie in its high chemoselectivity and its use of a cheap environmentally friendly metal catalyst. MPV reductions have been described as "obsolete" owing to the development of sodium borohydride and related reagents.
In organic chemistry, kinetic resolution is a means of differentiating two enantiomers in a racemic mixture. In kinetic resolution, two enantiomers react with different reaction rates in a chemical reaction with a chiral catalyst or reagent, resulting in an enantioenriched sample of the less reactive enantiomer. As opposed to chiral resolution, kinetic resolution does not rely on different physical properties of diastereomeric products, but rather on the different chemical properties of the racemic starting materials. The enantiomeric excess (ee) of the unreacted starting material continually rises as more product is formed, reaching 100% just before full completion of the reaction. Kinetic resolution relies upon differences in reactivity between enantiomers or enantiomeric complexes.
Oseltamivir total synthesis concerns the total synthesis of the antiinfluenza drug oseltamivir marketed by Hoffmann-La Roche under the trade name Tamiflu. Its commercial production starts from the biomolecule shikimic acid harvested from Chinese star anise and from recombinant E. coli. Control of stereochemistry is important: the molecule has three stereocenters and the sought-after isomer is only 1 of 8 stereoisomers.
Chiral Lewis acids (CLAs) are a type of Lewis acid catalyst. These acids affect the chirality of the substrate as they react with it. In such reactions, synthesis favors the formation of a specific enantiomer or diastereomer. The method is an enantioselective asymmetric synthesis reaction. Since they affect chirality, they produce optically active products from optically inactive or mixed starting materials. This type of preferential formation of one enantiomer or diastereomer over the other is formally known as asymmetric induction. In this kind of Lewis acid, the electron-accepting atom is typically a metal, such as indium, zinc, lithium, aluminium, titanium, or boron. The chiral-altering ligands employed for synthesizing these acids often have multiple Lewis basic sites that allow the formation of a ring structure involving the metal atom.
Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.
Cholesterol total synthesis in chemistry describes the total synthesis of the complex biomolecule cholesterol and is considered a great scientific achievement. The research group of Robert Robinson with John Cornforth published their synthesis in 1951 and that of Robert Burns Woodward with Franz Sondheimer in 1952. Both groups competed for the first publication since 1950 with Robinson having started in 1932 and Woodward in 1949. According to historian Greg Mulheirn the Robinson effort was hampered by his micromanagement style of leadership and the Woodward effort was greatly facilitated by his good relationships with chemical industry. Around 1949 steroids like cortisone were produced from natural resources but expensive. Chemical companies Merck & Co. and Monsanto saw commercial opportunities for steroid synthesis and not only funded Woodward but also provided him with large quantities of certain chemical intermediates from pilot plants. Hard work also helped the Woodward effort: one of the intermediate compounds was named Christmasterone as it was synthesized on Christmas Day 1950 by Sondheimer.
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.
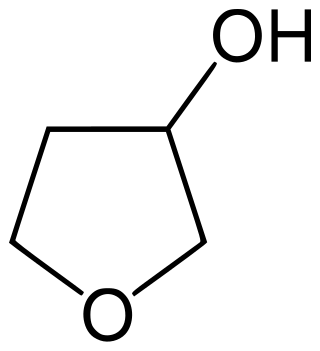
3-Hydroxytetrahydrofuran is a colorless liquid with a normal boiling point of 179 °C and boiling at 88−89 °C at 17 mmHg, with density. 3-OH THF is a useful pharmaceutical intermediate. The enantiopure version of this compound is an intermediate to launched retroviral drugs.
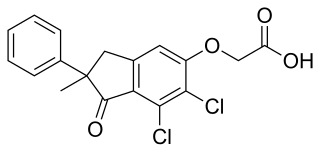
Indacrinone is a loop diuretic. It can be used in patients of gout with hypertension as an antihypertensive because it decreases reabsorption of uric acid, while other diuretics increase it.
Shiina esterification is an organic chemical reaction that synthesizes carboxylic esters from nearly equal amounts of carboxylic acids and alcohols by using aromatic carboxylic acid anhydrides as dehydration condensation agents. In 1994, Prof. Isamu Shiina reported an acidic coupling method using Lewis acid, and, in 2002, a basic esterification using nucleophilic catalyst.
The ketimine Mannich reaction is an asymmetric synthetic technique using differences in starting material to push a Mannich reaction to create an enantiomeric product with steric and electronic effects, through the creation of a ketimine group. Typically, this is done with a reaction with proline or another nitrogen-containing heterocycle, which control chirality with that of the catalyst. This has been theorized to be caused by the restriction of undesired (E)-isomer by preventing the ketone from accessing non-reactive tautomers. Generally, a Mannich reaction is the combination of an amine, a ketone with a β-acidic proton and aldehyde to create a condensed product in a β-addition to the ketone. This occurs through an attack on the ketone with a suitable catalytic-amine unto its electron-starved carbon, from which an imine is created. This then undergoes electrophilic addition with a compound containing an acidic proton. It is theoretically possible for either of the carbonyl-containing molecules to create diastereomers, but with the addition of catalysts which restrict addition as of the enamine creation, it is possible to extract a single product with limited purification steps and in some cases as reported by List et al.; practical one-pot syntheses are possible. The process of selecting a carbonyl-group gives the reaction a direct versus indirect distinction, wherein the latter case represents pre-formed products restricting the reaction's pathway and the other does not. Ketimines selects a reaction group, and circumvent a requirement for indirect pathways.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.