The Sonogashira reaction is a cross-coupling reaction used in organic synthesis to form carbon–carbon bonds. It employs a palladium catalyst as well as copper co-catalyst to form a carbon–carbon bond between a terminal alkyne and an aryl or vinyl halide.
An alkyne trimerisation is a [2+2+2] cycloaddition reaction in which three alkyne units react to form a benzene ring. The reaction requires a metal catalyst. The process is of historic interest as well as being applicable to organic synthesis. Being a cycloaddition reaction, it has high atom economy. Many variations have been developed, including cyclisation of mixtures of alkynes and alkenes as well as alkynes and nitriles.
Organopalladium chemistry is a branch of organometallic chemistry that deals with organic palladium compounds and their reactions. Palladium is often used as a catalyst in the reduction of alkenes and alkynes with hydrogen. This process involves the formation of a palladium-carbon covalent bond. Palladium is also prominent in carbon-carbon coupling reactions, as demonstrated in tandem reactions.
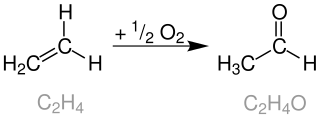
The Wacker process or the Hoechst-Wacker process refers to the oxidation of ethylene to acetaldehyde in the presence of palladium(II) chloride and copper(II) chloride as the catalyst. This chemical reaction was one of the first homogeneous catalysis with organopalladium chemistry applied on an industrial scale.
The Negishi coupling is a widely employed transition metal catalyzed cross-coupling reaction. The reaction couples organic halides or triflates with organozinc compounds, forming carbon-carbon bonds (C-C) in the process. A palladium (0) species is generally utilized as the metal catalyst, though nickel is sometimes used. A variety of nickel catalysts in either Ni0 or NiII oxidation state can be employed in Negishi cross couplings such as Ni(PPh3)4, Ni(acac)2, Ni(COD)2 etc.

Organocopper chemistry is the study of the physical properties, reactions, and synthesis of organocopper compounds, which are organometallic compounds containing a carbon to copper chemical bond. They are reagents in organic chemistry.

A boronic acid is an organic compound related to boric acid in which one of the three hydroxyl groups is replaced by an alkyl or aryl group. As a compound containing a carbon–boron bond, members of this class thus belong to the larger class of organoboranes.
A carbometallation is any reaction where a carbon-metal bond reacts with a carbon-carbon π-bond to produce a new carbon-carbon σ-bond and a carbon-metal σ-bond. The resulting carbon-metal bond can undergo further carbometallation reactions or it can be reacted with a variety of electrophiles including halogenating reagents, carbonyls, oxygen, and inorganic salts to produce different organometallic reagents. Carbometallations can be performed on alkynes and alkenes to form products with high geometric purity or enantioselectivity, respectively. Some metals prefer to give the anti-addition product with high selectivity and some yield the syn-addition product. The outcome of syn and anti- addition products is determined by the mechanism of the carbometallation.
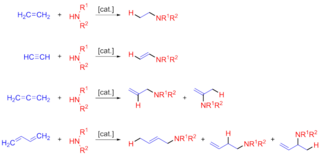
In organic chemistry, hydroamination is the addition of an N−H bond of an amine across a carbon-carbon multiple bond of an alkene, alkyne, diene, or allene. In the ideal case, hydroamination is atom economical and green. Amines are common in fine-chemical, pharmaceutical, and agricultural industries. Hydroamination can be used intramolecularly to create heterocycles or intermolecularly with a separate amine and unsaturated compound. The development of catalysts for hydroamination remains an active area, especially for alkenes. Although practical hydroamination reactions can be effected for dienes and electrophilic alkenes, the term hydroamination often implies reactions metal-catalyzed processes.
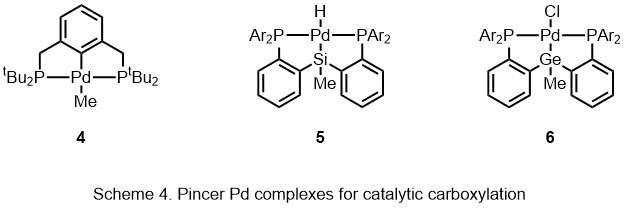
Carboxylation is a chemical reaction in which a carboxylic acid is produced by treating a substrate with carbon dioxide. The opposite reaction is decarboxylation. In chemistry, the term carbonation is sometimes used synonymously with carboxylation, especially when applied to the reaction of carbanionic reagents with CO2. More generally, carbonation usually describes the production of carbonates.

Organonickel chemistry is a branch of organometallic chemistry that deals with organic compounds featuring nickel-carbon bonds. They are used as a catalyst, as a building block in organic chemistry and in chemical vapor deposition. Organonickel compounds are also short-lived intermediates in organic reactions. The first organonickel compound was nickel tetracarbonyl Ni(CO)4, reported in 1890 and quickly applied in the Mond process for nickel purification. Organonickel complexes are prominent in numerous industrial processes including carbonylations, hydrocyanation, and the Shell higher olefin process.

The Liebeskind–Srogl coupling reaction is an organic reaction forming a new carbon–carbon bond from a thioester and a boronic acid using a metal catalyst. It is a cross-coupling reaction. This reaction was invented by and named after Jiri Srogl from the Academy of Sciences, Czech Republic, and Lanny S. Liebeskind from Emory University, Atlanta, Georgia, USA. There are three generations of this reaction, with the first generation shown below. The original transformation used catalytic Pd(0), TFP = tris(2-furyl)phosphine as an additional ligand and stoichiometric CuTC = copper(I) thiophene-2-carboxylate as a co-metal catalyst. The overall reaction scheme is shown below.

Organocobalt chemistry is the chemistry of organometallic compounds containing a carbon to cobalt chemical bond. Organocobalt compounds are involved in several organic reactions and the important biomolecule vitamin B12 has a cobalt-carbon bond. Many organocobalt compounds exhibit useful catalytic properties, the preeminent example being dicobalt octacarbonyl.
Organoplatinum chemistry is the chemistry of organometallic compounds containing a carbon to platinum chemical bond, and the study of platinum as a catalyst in organic reactions. Organoplatinum compounds exist in oxidation state 0 to IV, with oxidation state II most abundant. The general order in bond strength is Pt-C (sp) > Pt-O > Pt-N > Pt-C (sp3). Organoplatinum and organopalladium chemistry are similar, but organoplatinum compounds are more stable and therefore less useful as catalysts.
The Tsuji–Trost reaction is a palladium-catalysed substitution reaction involving a substrate that contains a leaving group in an allylic position. The palladium catalyst first coordinates with the allyl group and then undergoes oxidative addition, forming the π-allyl complex. This allyl complex can then be attacked by a nucleophile, resulting in the substituted product.

A metal-phosphine complex is a coordination complex containing one or more phosphine ligands. Almost always, the phosphine is an organophosphine of the type R3P (R = alkyl, aryl). Metal phosphine complexes are useful in homogeneous catalysis. Prominent examples of metal phosphine complexes include Wilkinson's catalyst (Rh(PPh3)3Cl), Grubbs' catalyst, and tetrakis(triphenylphosphine)palladium(0).
Decarboxylative cross coupling reactions are chemical reactions in which a carboxylic acid is reacted with an organic halide to form a new carbon-carbon bond, concomitant with loss of CO2. Aryl and alkyl halides participate. Metal catalyst, base, and oxidant are required.

In organometallic chemistry, the activation of cyclopropanes by transition metals is a research theme with implications for organic synthesis and homogeneous catalysis. Being highly strained, cyclopropanes are prone to oxidative addition to transition metal complexes. The resulting metallacycles are susceptible to a variety of reactions. These reactions are rare examples of C-C bond activation. The rarity of C-C activation processes has been attributed to Steric effects that protect C-C bonds. Furthermore, the directionality of C-C bonds as compared to C-H bonds makes orbital interaction with transition metals less favorable. Thermodynamically, C-C bond activation is more favored than C-H bond activation as the strength of a typical C-C bond is around 90 kcal per mole while the strength of a typical unactivated C-H bond is around 104 kcal per mole.
Heterobimetallic catalysis is an approach to catalysis that employs two different metals to promote a chemical reaction. Included in this definition are cases where: 1) each metal activates a different substrate, 2) both metals interact with the same substrate, and 3) only one metal directly interacts with the substrate(s), while the second metal interacts with the first.
In organic chemistry, carboboration describes an addition of both a carbon and a boron moiety to certain carbon-containing double and triple bonds, such as alkenes, alkynes, and allenes.