
Neurotoxins are toxins that are destructive to nerve tissue. Neurotoxins are an extensive class of exogenous chemical neurological insults that can adversely affect function in both developing and mature nervous tissue. The term can also be used to classify endogenous compounds, which, when abnormally contacted, can prove neurologically toxic. Though neurotoxins are often neurologically destructive, their ability to specifically target neural components is important in the study of nervous systems. Common examples of neurotoxins include lead, ethanol, glutamate, nitric oxide, botulinum toxin, tetanus toxin, and tetrodotoxin. Some substances such as nitric oxide and glutamate are in fact essential for proper function of the body and only exert neurotoxic effects at excessive concentrations.

N-methyl-D-aspartic acid or N-methyl-D-aspartate (NMDA) is an amino acid derivative that acts as a specific agonist at the NMDA receptor mimicking the action of glutamate, the neurotransmitter which normally acts at that receptor. Unlike glutamate, NMDA only binds to and regulates the NMDA receptor and has no effect on other glutamate receptors. NMDA receptors are particularly important when they become overactive during, for example, withdrawal from alcohol as this causes symptoms such as agitation and, sometimes, epileptiform seizures.

The N-methyl-D-aspartatereceptor (also known as the NMDA receptor or NMDAR), is a glutamate receptor and ion channel found in neurons. The NMDA receptor is one of three types of ionotropic glutamate receptors, the other two being AMPA and kainate receptors. Depending on its subunit composition, its ligands are glutamate and glycine (or D-serine). However, the binding of the ligands is typically not sufficient to open the channel as it may be blocked by Mg2+ ions which are only removed when the neuron is sufficiently depolarized. Thus, the channel acts as a “coincidence detector” and only once both of these conditions are met, the channel opens and it allows positively charged ions (cations) to flow through the cell membrane. The NMDA receptor is thought to be very important for controlling synaptic plasticity and mediating learning and memory functions.

An excitatory synapse is a synapse in which an action potential in a presynaptic neuron increases the probability of an action potential occurring in a postsynaptic cell. Neurons form networks through which nerve impulses travels, each neuron often making numerous connections with other cells of neurons. These electrical signals may be excitatory or inhibitory, and, if the total of excitatory influences exceeds that of the inhibitory influences, the neuron will generate a new action potential at its axon hillock, thus transmitting the information to yet another cell.

In excitotoxicity, nerve cells suffer damage or death when the levels of otherwise necessary and safe neurotransmitters such as glutamate become pathologically high, resulting in excessive stimulation of receptors. For example, when glutamate receptors such as the NMDA receptor or AMPA receptor encounter excessive levels of the excitatory neurotransmitter, glutamate, significant neuronal damage might ensue. Excess glutamate allows high levels of calcium ions (Ca2+) to enter the cell. Ca2+ influx into cells activates a number of enzymes, including phospholipases, endonucleases, and proteases such as calpain. These enzymes go on to damage cell structures such as components of the cytoskeleton, membrane, and DNA. In evolved, complex adaptive systems such as biological life it must be understood that mechanisms are rarely, if ever, simplistically direct. For example, NMDA in subtoxic amounts induces neuronal survival of otherwise toxic levels of glutamate.
Neuroprotection refers to the relative preservation of neuronal structure and/or function. In the case of an ongoing insult the relative preservation of neuronal integrity implies a reduction in the rate of neuronal loss over time, which can be expressed as a differential equation. It is a widely explored treatment option for many central nervous system (CNS) disorders including neurodegenerative diseases, stroke, traumatic brain injury, spinal cord injury, and acute management of neurotoxin consumption. Neuroprotection aims to prevent or slow disease progression and secondary injuries by halting or at least slowing the loss of neurons. Despite differences in symptoms or injuries associated with CNS disorders, many of the mechanisms behind neurodegeneration are the same. Common mechanisms of neuronal injury include decreased delivery of oxygen and glucose to the brain, energy failure, increased levels in oxidative stress, mitochondrial dysfunction, excitotoxicity, inflammatory changes, iron accumulation, and protein aggregation. Of these mechanisms, neuroprotective treatments often target oxidative stress and excitotoxicity—both of which are highly associated with CNS disorders. Not only can oxidative stress and excitotoxicity trigger neuron cell death but when combined they have synergistic effects that cause even more degradation than on their own. Thus limiting excitotoxicity and oxidative stress is a very important aspect of neuroprotection. Common neuroprotective treatments are glutamate antagonists and antioxidants, which aim to limit excitotoxicity and oxidative stress respectively.

Glutamate receptors are synaptic and non synaptic receptors located primarily on the membranes of neuronal and glial cells. Glutamate is abundant in the human body, but particularly in the nervous system and especially prominent in the human brain where it is the body's most prominent neurotransmitter, the brain's main excitatory neurotransmitter, and also the precursor for GABA, the brain's main inhibitory neurotransmitter. Glutamate receptors are responsible for the glutamate-mediated postsynaptic excitation of neural cells, and are important for neural communication, memory formation, learning, and regulation.
Glutamate transporters are a family of neurotransmitter transporter proteins that move glutamate – the principal excitatory neurotransmitter – across a membrane. The family of glutamate transporters is composed of two primary subclasses: the excitatory amino acid transporter (EAAT) family and vesicular glutamate transporter (VGLUT) family. In the brain, EAATs remove glutamate from the synaptic cleft and extrasynaptic sites via glutamate reuptake into glial cells and neurons, while VGLUTs move glutamate from the cell cytoplasm into synaptic vesicles. Glutamate transporters also transport aspartate and are present in virtually all peripheral tissues, including the heart, liver, testes, and bone. They exhibit stereoselectivity for L-glutamate but transport both L-aspartate and D-aspartate.

l-Kynurenine is a metabolite of the amino acid l-tryptophan used in the production of niacin.
Gliosis is a nonspecific reactive change of glial cells in response to damage to the central nervous system (CNS). In most cases, gliosis involves the proliferation or hypertrophy of several different types of glial cells, including astrocytes, microglia, and oligodendrocytes. In its most extreme form, the proliferation associated with gliosis leads to the formation of a glial scar.
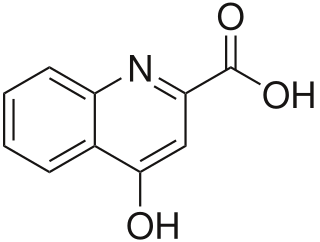
Kynurenic acid is a product of the normal metabolism of amino acid L-tryptophan. It has been shown that kynurenic acid possesses neuroactive activity. It acts as an antiexcitotoxic and anticonvulsant, most likely through acting as an antagonist at excitatory amino acid receptors. Because of this activity, it may influence important neurophysiological and neuropathological processes. As a result, kynurenic acid has been considered for use in therapy in certain neurobiological disorders. Conversely, increased levels of kynurenic acid have also been linked to certain pathological conditions.

NMDA receptor antagonists are a class of drugs that work to antagonize, or inhibit the action of, the N-Methyl-D-aspartate receptor (NMDAR). They are commonly used as anesthetics for animals and humans; the state of anesthesia they induce is referred to as dissociative anesthesia.
Gliotransmitters are chemicals released from glial cells that facilitate neuronal communication between neurons and other glial cells. They are usually induced from Ca2+ signaling, although recent research has questioned the role of Ca2+ in gliotransmitters and may require a revision of the relevance of gliotransmitters in neuronal signalling in general.

In enzymology, a kynurenine 3-monooxygenase (EC 1.14.13.9) is an enzyme that catalyzes the chemical reaction

Glutamate [NMDA] receptor subunit 3A is a protein that in humans is encoded by the GRIN3A gene.

Glutamate [NMDA] receptor subunit 3B is a protein that in humans is encoded by the GRIN3B gene.

Cystine/glutamate transporter is an antiporter that in humans is encoded by the SLC7A11 gene.

Hypertryptophanemia is a rare autosomal recessive metabolic disorder that results in a massive buildup of the amino acid tryptophan in the blood, with associated symptoms and tryptophanuria.
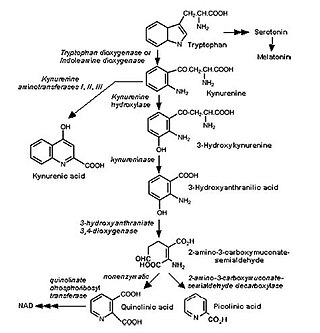
The kynurenine pathway is a metabolic pathway leading to the production of nicotinamide adenine dinucleotide (NAD+). Metabolites involved in the kynurenine pathway include tryptophan, kynurenine, kynurenic acid, xanthurenic acid, quinolinic acid, and 3-hydroxykynurenine. The kynurenine pathway is responsible for total catabolization of tryptophan about 95%. Disruption in the pathway is associated with certain genetic and psychiatric disorders.
Immuno-psychiatry, according to Pariante, is a discipline that studies the connection between the brain and the immune system. It differs from psychoneuroimmunology by postulating that behaviors and emotions are governed by peripheral immune mechanisms. Depression, for instance, is seen as malfunctioning of the immune system.


