Related Research Articles

A genetic disorder is a health problem caused by one or more abnormalities in the genome. It can be caused by a mutation in a single gene (monogenic) or multiple genes (polygenic) or by a chromosome abnormality. Although polygenic disorders are the most common, the term is mostly used when discussing disorders with a single genetic cause, either in a gene or chromosome. The mutation responsible can occur spontaneously before embryonic development, or it can be inherited from two parents who are carriers of a faulty gene or from a parent with the disorder. When the genetic disorder is inherited from one or both parents, it is also classified as a hereditary disease. Some disorders are caused by a mutation on the X chromosome and have X-linked inheritance. Very few disorders are inherited on the Y chromosome or mitochondrial DNA.

Joubert syndrome is a rare autosomal recessive genetic disorder that affects the cerebellum, an area of the brain that controls balance and coordination.
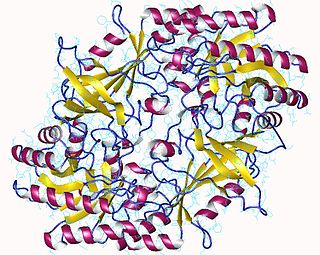
The enzyme ornithine decarboxylase catalyzes the decarboxylation of ornithine to form putrescine. This reaction is the committed step in polyamine synthesis. In humans, this protein has 461 amino acids and forms a homodimer.

Kabuki syndrome is a rare congenital disorder of genetic origin. It affects multiple parts of the body, with varying symptoms and severity, although the most common is the characteristic facial appearance.

Lesch–Nyhan syndrome (LNS) is a rare inherited disorder caused by a deficiency of the enzyme hypoxanthine-guanine phosphoribosyltransferase (HGPRT). This deficiency occurs due to mutations in the HPRT1 gene located on the X chromosome. LNS affects about 1 in 380,000 live births. The disorder was first recognized and clinically characterized by American medical student Michael Lesch and his mentor, pediatrician William Nyhan, at Johns Hopkins.
Albinism-black lock-cell migration disorder is the initialism for the following terms and concepts that describe a condition affecting a person's physical appearance and physiology: (1) A – albinism, (2) B – black lock of hair, (3) C – cell migration disorder of the neurocytes of the gut, and (4) D – sensorineural deafness. The syndrome is caused by mutation in the endothelin B receptor gene (EDNRB).

Argininosuccinic aciduria is an inherited disorder that causes the accumulation of argininosuccinic acid in the blood and urine. Some patients may also have an elevation of ammonia, a toxic chemical, which can affect the nervous system. Argininosuccinic aciduria may become evident in the first few days of life because of high blood ammonia, or later in life presenting with "sparse" or "brittle" hair, developmental delay, and tremors.

Alström syndrome (AS), also called Alström–Hallgren syndrome, is a very rare autosomal recessive genetic disorder characterised by childhood obesity and multiple organ dysfunction. Symptoms include early-onset type 2 diabetes, cone-rod dystrophy resulting in blindness, sensorineural hearing loss and dilated cardiomyopathy. Endocrine disorders typically also occur, such as hypergonadotrophic hypogonadism and hypothyroidism, as well as acanthosis nigricans resulting from hyperinsulinemia. Developmental delay is seen in almost half of people with Alström syndrome.
Conradi–Hünermann syndrome is a rare type of chondrodysplasia punctata. It is associated with the EBP gene and affects between one in 100,000 and one in 200,000 babies.
Rabson–Mendenhall syndrome is a rare autosomal recessive disorder characterized by severe insulin resistance. The disorder is caused by mutations in the insulin receptor gene. Symptoms include growth abnormalities of the head, face and nails, along with the development of acanthosis nigricans. Treatment involves controlling blood glucose levels by using insulin and incorporating a strategically planned, controlled diet. Also, direct actions against other symptoms may be taken This syndrome usually affects children and has a prognosis of 1–2 years.
Progressive Myoclonic Epilepsies (PME) are a rare group of inherited neurodegenerative diseases characterized by myoclonus, resistance to treatment, and neurological deterioration. The cause of PME depends largely on the type of PME. Most PMEs are caused by autosomal dominant or recessive and mitochondrial mutations. The location of the mutation also affects the inheritance and treatment of PME. Diagnosing PME is difficult due to their genetic heterogeneity and the lack of a genetic mutation identified in some patients. The prognosis depends largely on the worsening symptoms and failure to respond to treatment. There is no current cure for PME and treatment focuses on managing myoclonus and seizures through antiepileptic medication (AED).
3-M syndrome or 3M3 is a rare hereditary disorder characterized by severe growth retardation, facial dysmorphia, and skeletal abnormalities. The name 3-M is derived from the initials of the three researchers who first identified it: Miller, McKusick, and Malvaux and report their findings in the medical literature in 1972. Mutations in any one of the following three genes: CUL7, OBSL1, and CCDC8 are responsible for the occurrence of this disorder. It is inherited through an autosomal recessive pattern and considered very rare, so far less than 100 cases worldwide have been identified. Diagnosis is based on the presence of clinical features. Genetic testing can confirm the diagnosis and identify the specific gene involved. Treatment is aimed at addressing the growth and skeletal problems and may include surgical bone lengthening, adaptive aids, and physical therapy. An endocrinologist may assist with growth hormone replacement and appropriate evaluations during puberty.
Dopamine-responsive dystonia (DRD) also known as Segawa syndrome (SS), is a genetic movement disorder which usually manifests itself during early childhood at around ages 5–8 years.

Pitt–Hopkins syndrome (PTHS) is a rare genetic disorder characterized by developmental delay, moderate to severe intellectual disability, distinctive facial features, and possible intermittent hyperventilation followed by apnea. Epilepsy often occurs in Pitt-Hopkins. It is part of the clinical spectrum of Rett-like syndromes. Pitt-Hopkins syndrome is clinically similar to Angelman syndrome, Rett-syndrome, Mowat Wilson syndrome, and ATR-X syndrome.

Kufor–Rakeb syndrome (KRS) is an autosomal recessive disorder of juvenile onset also known as Parkinson disease-9 (PARK9). It is named after Kufr Rakeb in Irbid, Jordan. Kufor–Rakeb syndrome was first identified in this region in Jordan with a Jordanian couple's 5 children who had rigidity, mask-like face, and bradykinesia. The disease was first described in 1994 by Najim Al-Din et al. The OMIM number is 606693.

Cerebral folate deficiency is a condition in which concentrations of 5-methyltetrahydrofolate are low in the brain as measured in the cerebral spinal fluid despite being normal in the blood. Symptoms typically appear at about 5 to 24 months of age. Without treatment there may be poor muscle tone, trouble with coordination, trouble talking, and seizures.
Fryns-Aftimos syndrome is a rare chromosomal condition and is associated with pachygyria, severe mental retardation, epilepsy and characteristic facial features. This syndrome is a malformation syndrome, characterized by numerous facial dysmorphias not limited to hypertelorism, iris or retinal coloboma, cleft lip, and congenital heart defects. This syndrome has been seen in 30 unrelated people. Characterized by a de novo mutation located on chromosome 7p22, there is typically no family history prior to onset. The severity of the disorder can be determined by the size of the deletion on 7p22, enveloping the ACTB gene and surrounding genes, which is consistent with a contiguous gene deletion syndrome. Confirming a diagnosis of Fryns-Aftimos syndrome typically consists of serial single-gene testing or multigene panel of genes of interest or exome sequencing.

CDK13-related disorder, also known as congenital heart defects, dysmorphic facial features and intellectual developmental disorder (CHDFIDD), is a very rare autosomal dominant genetic condition characterised by congenital heart defects, intellectual disability and characteristic facial features. Those affected typically have motor and language delays, low muscle tone and gastrointestinal dysmotility. Facial features include a wide nasal bridge, widely-spaced eyes, prominent, low-set ears, a flat nose tip and a small mouth. Less common features include congenital spinal abnormalities, hearing loss or seizures.

SRD5A3-CDG is a rare, non X-linked congenital disorder of glycosylation (CDG) due to a mutation in the steroid 5 alpha reductase type 3 gene. It is one of over 150 documented types of Congenital disorders of Glycosylation. Like many other CDGs, SRD5A3 is ultra-rare, with around 38 documented cases in the world.

Severe intellectual disability-progressive spastic diplegia syndrome is a rare novel genetic disorder characterized by severe intellectual disabilities, ataxia, craniofacial dysmorphisms, and muscle spasticity. It is a type of autosomal dominant syndromic intellectual disability.
References
- 1 2 "Bachmann-Bupp Syndrome - Symptoms, Causes, Treatment | NORD". rarediseases.org. Retrieved 7 December 2023.
- 1 2 3 4 5 Bachmann, André S.; VanSickle, Elizabeth A.; Michael, Julianne; Vipond, Marlie; Bupp, Caleb P. (2024). "Bachmann-Bupp syndrome and treatment". Developmental Medicine and Child Neurology. 66 (4): 445–455. doi:10.1111/dmcn.15687. PMC 10796844 . PMID 37469105.
- 1 2 3 4 5 6 7 8 9 10 11 12 Bupp C, Michael J, VanSickle E, et al. Bachmann-Bupp Syndrome. 2022 Aug 25. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2023
- 1 2 Schultz, Chad R.; Bupp, Caleb P.; Rajasekaran, Surender; Bachmann, André S. (24 July 2019). "Biochemical features of primary cells from a pediatric patient with a gain-of-function ODC1 genetic mutation". Biochemical Journal. 476 (14): 2047–2057. doi:10.1042/bcj20190294. PMID 31249027.
- 1 2 "NIH awards MSU $4M to study rare children's disease Bachmann-Bupp Syndrome - MSU Innovation Center". MSU Innovation Center. 12 June 2023. Retrieved 23 December 2024.
- 1 2 3 4 5 6 Afrin, Antara; Afshan, Tonia S.; VanSickle, Elizabeth A.; Michael, Julianne; Laarman, Rachel L.; Bupp, Caleb P. (2023). "Improvement of dermatological symptoms in patients with Bachmann-Bupp syndrome using difluoromethylornithine treatment". Pediatric Dermatology. 40 (3): 528–531. doi:10.1111/pde.15187. PMID 36443247.
- 1 2 Heidt, Amanda (1 December 2021). "Doctors Treat Girl's Genetic Disorder with Repurposed Drug". The Scientist Magazine. Retrieved 23 December 2024.