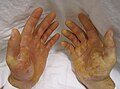
The therapy of genodermatosis not only needs to take care of patients' skin, reduce their pain and prevent complications, but also needs to carry out mental support for patients and their families. [5]
Prevention and care
Different types of genodermatosis require different kinds of prevention and care.
Ichthyosis
There is no radical therapeutic method for ichthyosis, but some care can ease the symptoms. [10] To prevent skin thickening and hydrate skin, patients can apply a cream containing alpha hydroxy acids and patients can also be treated with antibiotics for subsequent infections. [10]
Epidermolysis bullosa
For epidermolysis bullosa, daily care is important. When treating a wound, keep it clean and reduce friction, the bandages and dressing used must be non-sticky and gentle, and the patient should wear loose clothing to avoid damaging the wound. [13] [14]
Epidermolytic hyperkeratosis
The treatment of Epidermolytic hyperkeratosis is mainly control and alleviate symptoms, and good nursing can reduce the incidence of complications like electrolyte disturbances and sepsis. [18] To improve the look and feeling of the skin, patients can apply a cream containing alpha hydroxy acids, glycerol and urea, and if necessary, patients can use antibiotics to control secondary infections. [18]
Pachyonychia congenita
There is no radical therapeutic method for pachyonychia congenita, but some care can ease the symptoms. [16] Patients can polish and trim thickened nails, some of them can use retinoids to relieve symptoms but it may increase pain. [16]
Selumetinib and trametinib have been shown to reduce and control tumor growth in people with neurofibromatosis type I, reducing the likelihood of malignant lesions. [20]
Therapeutic methods
There are some new and developing genetic therapies available in genodermatosis. [20]
In the case of X-linked hypohidrotic ectodermal dysplasia, unborn babies diagnosed with this disease can be treated in their mothers' womb. Providing regulatory proteins in the womb during a critical period of infant growth may help correct the development of babies' sweat glands. [20]
Ustekinumab is a biologic therapy that can be used in a variety of genodermatosis such as congenital ichthyosis, psoriasis, deficiency of interleukin-36 receptor antagonist (DITRA) and so on. [20]
A new topical method could treat skin abnormalities in rare inherited lipid metabolic diseases. [21] This method obstructs abnormal mevalonate by topical application of lovastatin so that the production and cumulation of poisonous metabolic intermediates can be inhibited as much as possible and takes the place of the lacking lipid in the skin by topical application of cholesterol. [20] [21] The idea that similar treatments could be developed for other genodermatosis was also pointed out at the annual conference of the European Society of Dermatology and Venereology. [21]
For epidermolysis bullosa, a method called CRISPR can be used to treat this disease by gene analysis, modification and substitution, but the method is ethically controversial because it consents to editing genes highly. [20]
Treatments for some rare diseases are still being studied. The therapy of genodermatosis requires the updating of technology, and the development of technology depends on the continuous understanding of the mechanism of the disease. Research on the treatment of genodermatosis is at a positive stage of development. [20]