
A thymoma is a tumor originating from the epithelial cells of the thymus that is considered a rare malignancy. Thymomas are frequently associated with neuromuscular disorders such as myasthenia gravis; thymoma is found in 20% of patients with myasthenia gravis. Once diagnosed, thymomas may be removed surgically. In the rare case of a malignant tumor, chemotherapy may be used.

Hemangioendotheliomas are a family of vascular neoplasms of intermediate malignancy.
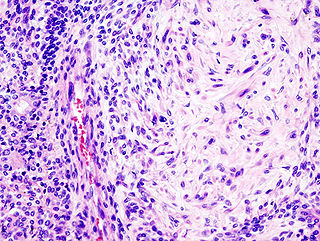
Pleomorphic adenoma is a common benign salivary gland neoplasm characterised by neoplastic proliferation of parenchymatous glandular cells along with myoepithelial components, having a malignant potentiality. It is the most common type of salivary gland tumor and the most common tumor of the parotid gland. It derives its name from the architectural Pleomorphism seen by light microscopy. It is also known as "Mixed tumor, salivary gland type", which refers to its dual origin from epithelial and myoepithelial elements as opposed to its pleomorphic appearance.

Hürthle cell adenoma is a rare benign tumor, typically seen in women between the ages of 70 and 80 years old. This adenoma is characterized by a mass of benign Hürthle cells. Typically such a mass is removed because it is not easy to predict whether it will transform into the malignant counterpart, a subtype of follicular thyroid cancer called a Hürthle cell carcinoma.

Canalicular adenoma is a benign, epithelial salivary gland neoplasm arranged in interconnecting cords of columnar cells. This is a very rare benign neoplasm, that makes up about 1% of all salivary gland tumors, or about 4% of all benign salivary gland tumors.

An atypical teratoid rhabdoid tumor (AT/RT) is a rare tumor usually diagnosed in childhood. Although usually a brain tumor, AT/RT can occur anywhere in the central nervous system (CNS), including the spinal cord. About 60% will be in the posterior cranial fossa. One review estimated 52% in the posterior fossa, 39% are supratentorial primitive neuroectodermal tumors (sPNET), 5% are in the pineal, 2% are spinal, and 2% are multifocal.
Alveolar rhabdomyosarcoma (ARMS) is a subtype of the rhabdomyosarcoma soft tissue cancer family whose lineage is from mesenchymal cells and are related to skeletal muscle cells. ARMS tumors resemble the alveolar tissue in the lungs. Tumor location varies from patient to patient, but is commonly found in the head and neck region, male and female urogenital tracts, the torso, and extremities. Two fusion proteins can be associated with ARMS, but are not necessary, PAX3-FKHR. and PAX7-FKHR. In children and adolescents ARMS accounts for about 1 percent of all malignancies, has an incidence rate of 1 per million, and most cases occur sporadically with no genetic predisposition. PAX3-FOXO1 is now known to drive cancer-promoting gene expression programs through creation of distant genetic elements called super enhancers.
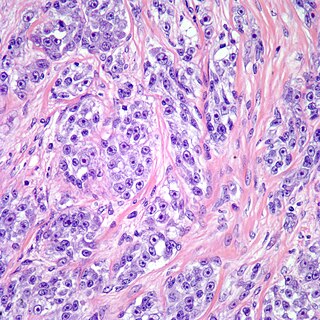
Clear cell sarcoma is a rare form of cancer called a sarcoma. It is known to occur mainly in the soft tissues and dermis. Rare forms were thought to occur in the gastrointestinal tract before they were discovered to be different and redesignated as GNET.
A mixed tumor is a tumor that derives from multiple tissue types. A biplastic tumor or biphasic tumor has two tissue types.
Oral pigmentation is asymptomatic and does not usually cause any alteration to the texture or thickness of the affected area. The colour can be uniform or speckled and can appear solitary or as multiple lesions. Depending on the site, depth, and quantity of pigment, the appearance can vary considerably.
Large cell lung carcinoma with rhabdoid phenotype (LCLC-RP) is a rare histological form of lung cancer, currently classified as a variant of large cell lung carcinoma (LCLC). In order for a LCLC to be subclassified as the rhabdoid phenotype variant, at least 10% of the malignant tumor cells must contain distinctive structures composed of tangled intermediate filaments that displace the cell nucleus outward toward the cell membrane. The whorled eosinophilic inclusions in LCLC-RP cells give it a microscopic resemblance to malignant cells found in rhabdomyosarcoma (RMS), a rare neoplasm arising from transformed skeletal muscle. Despite their microscopic similarities, LCLC-RP is not associated with rhabdomyosarcoma.
Mucinous cystadenocarcinoma of the lung (MCACL) is a very rare malignant mucus-producing neoplasm arising from the uncontrolled growth of transformed epithelial cells originating in lung tissue.
Skin cancer, or neoplasia, is the most common type of cancer diagnosed in horses, accounting for 45 to 80% of all cancers diagnosed. Sarcoids are the most common type of skin neoplasm and are the most common type of cancer overall in horses. Squamous-cell carcinoma is the second-most prevalent skin cancer, followed by melanoma. Squamous-cell carcinoma and melanoma usually occur in horses greater than 9-years-old, while sarcoids commonly affect horses 3 to 6 years old. Surgical biopsy is the method of choice for diagnosis of most equine skin cancers, but is contraindicated for cases of sarcoids. Prognosis and treatment effectiveness varies based on type of cancer, degree of local tissue destruction, evidence of spread to other organs (metastasis) and location of the tumor. Not all cancers metastasize and some can be cured or mitigated by surgical removal of the cancerous tissue or through use of chemotherapeutic drugs.
A sialoblastoma is a low-grade salivary gland neoplasm that recapitulates primitive salivary gland anlage. It has previously been referred to as congenital basal cell adenoma, embryoma, or basaloid adenocarcinoma. It is an extremely rare tumor, with less than 100 cases reported worldwide.
Rhabdomyoma is a benign mesenchymal tumor of skeletal muscle, separated into two major categories based on site: Cardiac and extracardiac. They are further separated by histology: fetal, juvenile (intermediate), and adult types. Genital types are recognized, but are often part of either the fetal or juvenile types. The fetal type is thought to recapitulate immature skeletal muscle at about week six to ten of gestational development.
A ceruminous adenoma is a benign glandular neoplasm which arises from the ceruminous glands located within the external auditory canal. These glands are found within the outer one third to one half of the external auditory canal, more common along the posterior surface; therefore, the tumor develops within a very specific location.
Neuroendocrine adenoma middle ear (NAME) is a tumor which arises from a specific anatomic site: middle ear. NAME is a benign glandular neoplasm of middle ear showing histologic and immunohistochemical neuroendocrine and mucin-secreting differentiation.

Sclerosing polycystic adenosis is a rare salivary gland tumor first described in 1996 by Dr. Brion Smith. The major salivary glands, specifically the parotid gland and the submandibular gland, are affected most commonly. Patients usually come to clinical attention with a mass or swelling in their salivary glands in the 5th decade of life, with females affected much more commonly than males. Nearly all of the cases reported so far have a benign behavior, although there is a single case that has had an associated malignant transformation.

Biphenotypic sinonasal sarcoma is a newly recognized, very rare, low-grade malignant tumor of the nasal cavity, which was formerly probably included in fibrosarcoma and synovial sarcoma cases. It was incorporated into the fourth edition of the World Health Organization Classification of Head and Neck Tumours, published in 2017.
Ectomesenchymal chondromyxoid tumor (ECT) is a benign intraoral tumor with presumed origin from undifferentiated (ecto)mesenchymal cells. There are some who think it is a myoepithelial tumor type.


