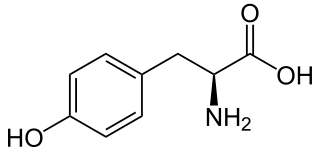
L-Tyrosine or tyrosine or 4-hydroxyphenylalanine is one of the 20 standard amino acids that are used by cells to synthesize proteins. It is a non-essential amino acid with a polar side group. The word "tyrosine" is from the Greek tyrós, meaning cheese, as it was first discovered in 1846 by German chemist Justus von Liebig in the protein casein from cheese. It is called tyrosyl when referred to as a functional group or side chain. While tyrosine is generally classified as a hydrophobic amino acid, it is more hydrophilic than phenylalanine. It is encoded by the codons UAC and UAU in messenger RNA.
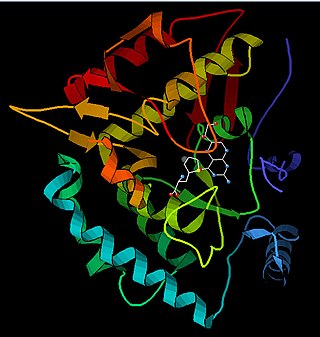
Phenylalanine hydroxylase. (PAH) (EC 1.14.16.1) is an enzyme that catalyzes the hydroxylation of the aromatic side-chain of phenylalanine to generate tyrosine. PAH is one of three members of the biopterin-dependent aromatic amino acid hydroxylases, a class of monooxygenase that uses tetrahydrobiopterin (BH4, a pteridine cofactor) and a non-heme iron for catalysis. During the reaction, molecular oxygen is heterolytically cleaved with sequential incorporation of one oxygen atom into BH4 and phenylalanine substrate. In humans, mutations in its encoding gene, PAH, can lead to the metabolic disorder phenylketonuria.
Isomerases are a general class of enzymes that convert a molecule from one isomer to another. Isomerases facilitate intramolecular rearrangements in which bonds are broken and formed. The general form of such a reaction is as follows:

A transferase is any one of a class of enzymes that catalyse the transfer of specific functional groups from one molecule to another. They are involved in hundreds of different biochemical pathways throughout biology, and are integral to some of life's most important processes.

Glutathione S-transferases (GSTs), previously known as ligandins, are a family of eukaryotic and prokaryotic phase II metabolic isozymes best known for their ability to catalyze the conjugation of the reduced form of glutathione (GSH) to xenobiotic substrates for the purpose of detoxification. The GST family consists of three superfamilies: the cytosolic, mitochondrial, and microsomal—also known as MAPEG—proteins. Members of the GST superfamily are extremely diverse in amino acid sequence, and a large fraction of the sequences deposited in public databases are of unknown function. The Enzyme Function Initiative (EFI) is using GSTs as a model superfamily to identify new GST functions.

Tyrosinemia or tyrosinaemia is an error of metabolism, usually inborn, in which the body cannot effectively break down the amino acid tyrosine. Symptoms of untreated tyrosinemia include liver and kidney disturbances. Without treatment, tyrosinemia leads to liver failure. Today, tyrosinemia is increasingly detected on newborn screening tests before any symptoms appear. With early and lifelong management involving a low-protein diet, special protein formula, and sometimes medication, people with tyrosinemia develop normally, are healthy, and live normal lives.

Homogentisic acid is a phenolic acid usually found in Arbutus unedo (strawberry-tree) honey. It is also present in the bacterial plant pathogen Xanthomonas campestris pv. phaseoli as well as in the yeast Yarrowia lipolytica where it is associated with the production of brown pigments. It is oxidatively dimerised to form hipposudoric acid, one of the main constituents of the 'blood sweat' of hippopotamuses.

4-Hydroxyphenylpyruvate dioxygenase (HPPD), also known as α-ketoisocaproate dioxygenase, is an Fe(II)-containing non-heme oxygenase that catalyzes the second reaction in the catabolism of tyrosine - the conversion of 4-hydroxyphenylpyruvate into homogentisate. HPPD also catalyzes the conversion of phenylpyruvate to 2-hydroxyphenylacetate and the conversion of α-ketoisocaproate to β-hydroxy β-methylbutyrate. HPPD is an enzyme that is found in nearly all aerobic forms of life.

4-Maleylacetoacetate is an intermediate in the metabolism of tyrosine. It is converted to fumarylacetoacetate by the enzyme 4-maleylacetoacetate cis-trans-isomerase. Gluthathione coenzymatically helps in conversion to fumarylacetoacetic acid.

Fumarylacetoacetic acid (fumarylacetoacetate) is an intermediate in the metabolism of tyrosine. It is formed through the conversion of maleylacetoacetate into fumarylacetoacetate by the enzyme maleylacetoacetate isomerase.

Fumarylacetoacetase is an enzyme that in humans is encoded by the FAH gene located on chromosome 15. The FAH gene is thought to be involved in the catabolism of the amino acid phenylalanine in humans.

In enzymology, a microsomal epoxide hydrolase (mEH) is an enzyme that catalyzes the hydrolysis reaction between an epoxide and water to form a diol.
The enzyme indole-3-glycerol-phosphate synthase (IGPS) (EC 4.1.1.48) catalyzes the chemical reaction

Glutathione S-transferase, C-terminal domain is a structural domain of glutathione S-transferase (GST).

Glutathione S-transferase Zeta 1 is an enzyme that in humans is encoded by the GSTZ1 gene on chromosome 14.
3-oxoacid CoA-transferase 1 (OXCT1) is an enzyme that in humans is encoded by the OXCT1 gene. It is also known as succinyl-CoA-3-oxaloacid CoA transferase (SCOT). Mutations in the OXCT1 gene are associated with succinyl-CoA:3-oxoacid CoA transferase deficiency. This gene encodes a member of the 3-oxoacid CoA-transferase gene family. The encoded protein is a homodimeric mitochondrial matrix enzyme that plays a central role in extrahepatic ketone body catabolism by catalyzing the reversible transfer of coenzyme A (CoA) from succinyl-CoA to acetoacetate.

Glutathione S-transferase A3 is an enzyme that in humans is encoded by the GSTA3 gene.

Bacterial glutathione transferases are part of a superfamily of enzymes that play a crucial role in cellular detoxification. The primary role of GSTs is to catalyze the conjugation of glutathione (GSH) with the electrophilic centers of a wide variety of molecules. The most commonly known substrates of GSTs are xenobiotic synthetic chemicals. There are also classes of GSTs that utilize glutathione as a cofactor rather than a substrate. Often these GSTs are involved in reduction of reactive oxidative species toxic to the bacterium. Conjugation with glutathione receptors reders toxic substances more soluble, and therefore more readily exocytosed from the cell.

Tyrosinemia type I is a genetic disorder that disrupts the metabolism of the amino acid tyrosine, resulting in damage primarily to the liver along with the kidneys and peripheral nerves. The inability of cells to process tyrosine can lead to chronic liver damage ending in liver failure, as well as renal disease and rickets. Symptoms such as poor growth and enlarged liver are associated with the clinical presentation of the disease. Clinical manifestation of disease occurs typically within the first two years of life. The severity of the disease is correlated with the timing of onset of symptoms, earlier being more severe.
Coenzyme A transferases (CoA-transferases) are transferase enzymes that catalyze the transfer of a coenzyme A group from an acyl-CoA donor to a carboxylic acid acceptor. Among other roles, they are responsible for transfer of CoA groups during fermentation and metabolism of ketone bodies. These enzymes are found in all three domains of life.






