The action of this substance involves inhibiting the proliferation of cancer cells and inducing their differentiation or apoptosis, although the exact mechanism of the drug's action is not fully understood. Arsenic, due to its toxic properties, had been used for centuries as an effective and virtually undetectable[3] poison for the senses.[4] In the 20th century, its anticancer properties were observed, but attempts at oral administration were unsuccessful. Only intravenous administration of the substance yielded positive results, particularly in the treatment of a rare form of cancer – acute promyelocytic leukemia. This therapy is introduced after failure of treatment with retinoids and chemotherapy.[5] The treatment is characterized by relative safety and few side effects. Research is ongoing to find other uses for the drug.
History
In the 18th century, William Withering discovered that arsenic trioxide, when used in small doses, exhibited therapeutic effects.[6] During the same period, Thomas Fowler prepared a 1% solution of arsenic and potassium carbonate, which was used to treat skin diseases (primarily psoriasis) until the 20th century.[7] An arsenic-based drug, arsphenamine, was also developed for the treatment of syphilis, synthesized by Paul Ehrlich, though it was eventually replaced by penicillin.[8] Arsenic compounds were widely used to treat various diseases in the 19th and early 20th centuries.[7][9][10]
The first reports of the anticancer activity of arsenic trioxide date back to 1878, when a report from Boston City Hospital described Fowler's solution lowering leukocyte levels in the blood of two healthy individuals and one patient.[4][11] Arsenic trioxide continued to be used in the treatment of leukemia until the introduction of radiotherapy. It made a resurgence in the 1930s when the first studies confirmed the high efficacy of arsenic trioxide in treating chronic myelogenous leukemia.[12]
In the late 1960s, physicians working at the Harbin Medical Academy in China were sent to a center focusing on traditional Chinese medicine, where they used a melanoma ointment, with arsenic as its main ingredient. At that time, the arsenal of anticancer drugs was limited, prompting doctors to experiment with arsenic. Early trials used oral administration, but it showed strong toxic effects. In March 1971, the first trials of intravenous arsenic began, which showed significantly lower toxicity. For many years, arsenic trioxide was administered to patients with various cancers, showing the best results in the treatment of acute promyelocytic leukemia.[13] More than half of the patients from the first trial in Harbin survived for five years, prompting further research across other centers in China,[14][15] and eventually at the Sloan-Kettering Memorial Institute in New York.[16] The results of clinical trials were favorable enough that in 2000, the drug received FDA approval.[17]
Mechanism of action
Anti-apoptotic protein Bcl-2 (isoform 1)AIF factor – one of the "death proteins"APAF-1 – one of the "death proteins" (illustrated in relation to ATP)Bak – one of the pro-apoptotic proteinsBax – one of the pro-apoptotic proteinsTranscription factor NF-κB (illustrated in relation to DNA)JNK kinase
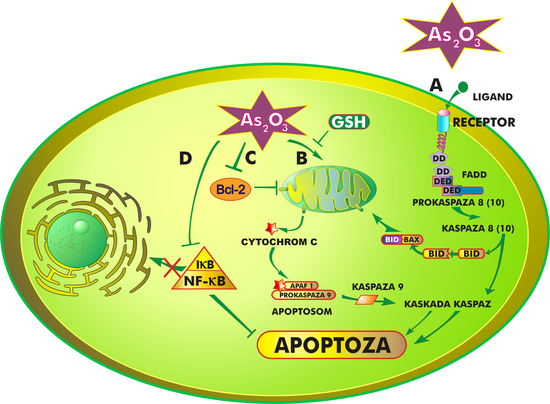
The mechanism of action of arsenic trioxide is complex and not fully understood. Generally, the drug inhibits the proliferation of cancer cells and induces their differentiation and/or apoptosis, which can occur in various ways depending on the involved organelles and biochemical processes. Arsenic trioxide induces apoptosis through:
Interaction with cell membrane receptors (extrinsic pathway)
The first of these pathways involves the binding of a ligand to a receptor located on the surface of the cell membrane. The interaction of these two entities leads to the activation of various genes and releases a cascade of proteins characteristic of the apoptosis process.[25]
Arsenic trioxide also interacts with mitochondria. One of the initial changes in their structure induced by the drug is the opening of megachannels and the release of so-called "death proteins", primarily cytochrome c, APAF-1 (apoptotic peptidase activating factor 1), AIF (apoptosis-inducing factor), Smac/DIABLO protein, and endonucleases from the intermembrane space of mitochondria into the cytosol. In the cytoplasm, a protein complex known as the apoptosome forms, which activates further processes leading to apoptosis.[26]
Regardless of whether apoptosis is induced externally or internally, it always involves caspases, whose activation irreversibly leads the cell down the path of programmed cell death.[27][28] Additionally, apoptosis is regulated by proteins from the Bcl-2 family, which can act as either pro-apoptotic or anti-apoptotic factors.[29]
The cause of acute promyelocytic leukemia is the translocation of the gene encoding the retinoic acid receptor (RARα) from chromosome 17 to a location near the PML gene on chromosome 15. This leads to the fusion of genes and the production of the PML/RARα protein.[30] This protein inhibits differentiation and the death of the cells in which it is present. Arsenic trioxide, even at low concentrations, causes the degradation of PML/RARα, thereby partially restoring the differentiation of cancerous promyelocytes.[31]
Arsenic trioxide activates JNK (c-Jun N-terminal kinase), also known as stress-activated protein kinase, which belongs to the MAPK (mitogen-activated protein kinase) family. These enzymes play a crucial role in signal transduction within the cell. Under normal conditions, JNK is activated by the phosphorylation of threonine and tyrosine residues.[32][33] However, studies on specific cell lines derived from patients with acute promyelocytic leukemia have demonstrated that this activation also occurs in response to arsenic trioxide.[34]
It seems that the activation of JNK leads to the phosphorylation of both anti-apoptotic proteins (Bcl-2, Bcl-Xl) and pro-apoptotic proteins – Bax (Bcl-2-associated X protein), Bak (Bcl-2 homologous killer), and Bid (BH3 interacting domain death agonist) – effectively activating them. Pro-apoptotic proteins contain the BH3 domain, which is responsible for their "death-inducing" activity. They cause the formation of ion channels in the mitochondrial membrane, resulting in the release of the aforementioned apoptotic factors into the cytoplasm. Anti-apoptotic proteins owe their function to a hydrophobic cleft in their spatial structure that binds to the BH3 domain, thereby neutralizing the effects of the "death" proteins.[35] Under normal conditions, the decision for a cell to undergo apoptosis depends on the ratio of pro-apoptotic to anti-apoptotic proteins. In the case of arsenic trioxide-induced apoptosis, two mechanisms play a significant role in increasing the levels of pro-apoptotic proteins. The first is related to the functioning of the transcription factorNF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells). NF-κB exists in the cytoplasm in an inactive state, in a complex with the specific reaction inhibitor IκB (IKK). This complex consists of two catalytic subunits – IKKα and IKKβ – and a regulatory unit IKKγ/NEMO. The phosphorylation and degradation of the inhibitor release NF-κB, which then translocates to the cell nucleus and activates genes responsible for producing "survival" proteins (such as p53, Bcl-2, and other inhibitors of apoptosis). NF-κB also protects cells from apoptotic stimulation involving the TNF-α receptor. Arsenic trioxide binds to the cysteine at position 179 of IKKβ, thus preventing the release of NF-κB.[35] The absence of this protein in the cytoplasm allows for the induction of apoptosis via the extrinsic pathway and activates caspases 3 and 8.[36]
This mechanism has been observed not only in acute promyelocytic leukemia cells and Hodgkin lymphoma but also in patients with myelodysplastic syndrome.[35][37][38] The second mechanism that increases the levels of pro-apoptotic proteins is the downregulation of bcl-2 gene transcription.[39] This effect has been observed in HL-60 and NB4 human leukemia cells.[40][41]
In 2003, Japanese researchers discovered that arsenic trioxide induces apoptosis not only through the TNF-α receptor. Studies indicate that the drug also acts pro-apoptotically through the CD95 receptor, which affects the activation of caspases 8 and 3.[42][43] In multiple myeloma cells, arsenic trioxide interacts with the APO2/TRAIL receptor, activating caspases 8 and 9.[44][45]
Arsenic trioxide also affects the intracellular concentration of glutathione, which is a crucial component of the redox system (it removes radicals and reduces hydrogen peroxide). It also participates, along with peroxidase and catalase, in regulating the levels of reactive oxygen species.[46] Arsenic trioxide inhibits glutathione peroxidase, thereby decreasing its concentration in the cell, which leads to an increase in the levels of reactive oxygen species.[47] These, in turn, increase the permeability of the mitochondrial membrane, causing the release of apoptotic factors and initiating the apoptosis process.[48]
Additionally, arsenic trioxide degrades poly(ADP-ribose) polymerase, which, combined with the activation of caspases, inhibits DNA repair and halts the cell cycle.[49] The phase of the cell cycle at which the blockage occurs primarily depends on the p53 protein. In cells containing the so-called "wild type" (non-mutated) p53, the cell cycle is halted in the interphase, while in cells with mutated p53, it is halted in the G2/M phase.[44][50]
Mechanism of action of arsenic trioxide. The illustration presents four main therapeutic pathways of arsenic trioxide: A – external apoptosis pathway, B – mitochondrial pathway, C – interaction of arsenic trioxide with Bcl-2 family proteins, D – impact of arsenic trioxide on the NF-κB transcription factor.
Clinical studies
Arsenic trioxide was clinically tested in two open-label, single-arm trials without a control group, involving 52 patients with acute promyelocytic leukemia who had previously been unsuccessfully treated with anthracyclines and retinoids. The results of these studies are presented in the table below.[52][53][54]
Study
Single-center n=12
Multi-center n=40
Dose of arsenic trioxide (mg/kg body weight/day)
0.16 (median: 0.06–0.20)
0.15
Complete remission
11 patients (92%)
34 patients (85%)
Average time to bone marrow remission
32 days
35 days
Average time to achieve complete remission
54 days
59 days
18-month survival rate
67%
66%
n – number of patients participating in the study
Studies have also been conducted on the effect of arsenic trioxide on other cancers. These showed that the drug also induces apoptosis in lung cancer cells (especially in combination with sulindac).[55] The efficacy of arsenic trioxide has also been demonstrated in the treatment of multiple myeloma, in combination with ascorbic acid[56] and bortezomib.[57]
Animal studies have shown that the drug also affects ovarian,[58] liver, stomach,[59] prostate, and breast cancers,[60] as well as gliomas[61] and pancreatic cancer (in combination with parthenolide).[62] However, attempts to use arsenic trioxide in the treatment of solid tumors have been limited by the drug's toxicity.[63]
Arsenic trioxide also appears promising for treating autoimmune diseases (based on studies in mice).[64]
Pharmacokinetics
Detailed pharmacokinetic studies on arsenic trioxide have not been conducted. When administered intravenously, a steady state is reached after 8–10 days. Arsenic binds to proteins to an insignificant extent. The highest concentrations of arsenic are found in the liver, kidneys, heart, lungs, hair, and nails. Arsenous acid is oxidized to arsenic acid and methylated in the liver,[65][66][67] and then excreted 60% in the urine. The drug has a half-life of 92 hours. Arsenic trioxide is neither a substrate nor an inhibitor of cytochrome P450isozymes (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5, 4A9/11).[68][69]
Indications
Arsenic trioxide is intended for the induction of remission and consolidation in adult patients with acute promyelocytic leukemia who have the t(15;17) translocation and/or the PML/RARα gene. The drug should be used after treatment failure or relapse. Prior therapy should include retinoid and chemotherapy.[70]
Special warnings
To ensure the safe use of arsenic trioxide, the following precautions should be observed:[71]
25% of patients treated with arsenic trioxide exhibited symptoms resembling leukocyte activation syndrome, characterized by high fever, shortness of breath, weight gain, pulmonary infiltrates with pleural or pericardialexudation, with or without leukocytosis. High doses of steroids (10mg dexamethasone intravenously, 2–3 times per day) appear to alleviate these symptoms.[72]
40% of patients treated with arsenic trioxide experienced at least one instance of prolonged QT interval, corrected to over 500 ms.[73] QT interval prolongation can lead to ventricular arrhythmias, such as torsades de pointes.[74]
Prior to initiating arsenic trioxide treatment, an ECG should be performed, and blood levels of potassium, calcium, magnesium, and creatinine should be checked. Any abnormalities, particularly a prolonged QT interval on the ECG, should be corrected before starting arsenic trioxide. Any medications that may prolong the QT interval should be discontinued if possible.[75]
Patients receiving arsenic trioxide, particularly those at risk for torsades de pointes, should be closely monitored during treatment.[76][77]
If toxicity reaches level 3 (as per National Cancer Institute criteria), treatment should be modified or discontinued before the planned completion of therapy. Patients can resume treatment only after symptoms subside, starting with 50% of the prior daily dose. The dose can be increased to the previous level if no toxicity symptoms appear within 3 days. If toxicity reappears, treatment with arsenic trioxide cannot continue.[71]
During the induction phase, electrolyte levels, glucose, blood counts, and liver and kidney function should be tested twice a week. In the consolidation phase, these tests should be performed weekly.[71]
During arsenic trioxide treatment, women of childbearing age and men capable of fathering children should use effective contraception. The impact of arsenic trioxide on fertility has not been thoroughly studied.[71]
Interactions
Arsenic trioxide is known to prolong the QT interval. If possible, medications that also prolong QT should not be used concurrently, including:[79][80]
Side effects were reported in 37% of patients treated with arsenic trioxide. However, these effects were generally mild and resolved during treatment. Patients tolerated consolidation therapy better than induction therapy. The most common side effects include:[81][82][83]
Other side effects include allergic skin reactions (including reactions at the injection site, injection site pain),[65]gastrointestinal disturbances (diarrhea), various types of pain, visual disturbances,[84] and bleeding.[85] If the drug extravasates, local irritation and phlebitis may occur.[65]
Overdose
In the event of arsenic poisoning (manifesting as seizures, muscle weakness, confusion),[86] the administration of the drug should be immediately discontinued, and appropriate treatment should be initiated. Penicillamine is commonly used at a dose of up to 1 g/day.[87] For patients unable to take oral medications, dimercaprol can be administered intramuscularly at a dose of 3mg/kg body weight every 4 hours[88] until life-threatening symptoms subside. In cases of coagulopathy,[89]DMSA is recommended at a dose of 10mg/kg body weight every 8 hours for 5 days, followed by every 12 hours for 2 weeks.[90]Kidney dialysis may also be considered.[91]
Preparations and form of the drug
Trisenox – Almac Pharma – 1mg/ml concentrate for intravenous therapy. Trisenox is packaged in 10 ml ampoules for single use.[92] The ampoules contain a pure solution of arsenic trioxide, without preservatives, and also contain sodium hydroxide and hydrochloric acid.[92] The solution has a pH of 7–9.[92] The drug should be stored at room temperature and must not be frozen.[86] After withdrawing the solution from the ampoule, it should be diluted in 100–250 ml of 5% glucose or saline solution. Arsenic trioxide should not be mixed or administered in the same infusion with other medications.[92]
In pharmaceutical compounding, arsenic trioxide was used in a 1:10 trituration with lactose (Trituratio Acidi arsenicosi 1/10). To prepare the trituration, one part arsenic trioxide is placed in a mortar, and while continuously grinding, nine parts of lactose are added in portions. Achieving a uniformly distributed trituration requires at least 10 minutes of grinding in the mortar.[1]
Related Research Articles
Apoptosis is a form of programmed cell death that occurs in multicellular organisms and in some eukaryotic, single-celled microorganisms such as yeast. Biochemical events lead to characteristic cell changes (morphology) and death. These changes include blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, DNA fragmentation, and mRNA decay. The average adult human loses 50 to 70 billion cells each day due to apoptosis. For the average human child between 8 and 14 years old, each day the approximate loss is 20 to 30 billion cells.
The Philadelphia chromosome or Philadelphia translocation (Ph) is a specific genetic abnormality in chromosome 22 of leukemia cancer cells. This chromosome is defective and unusually short because of reciprocal translocation, t(9;22)(q34;q11), of genetic material between chromosome 9 and chromosome 22, and contains a fusion gene called BCR-ABL1. This gene is the ABL1 gene of chromosome 9 juxtaposed onto the breakpoint cluster region BCR gene of chromosome 22, coding for a hybrid protein: a tyrosine kinase signaling protein that is "always on", causing the cell to divide uncontrollably by interrupting the stability of the genome and impairing various signaling pathways governing the cell cycle.
Bcl-2, encoded in humans by the BCL2 gene, is the founding member of the Bcl-2 family of regulator proteins. BCL2 blocks programmed cell death (apoptosis) while other BCL2 family members can either inhibit or induce it. It was the first apoptosis regulator identified in any organism.
Karyorrhexis is the destructive fragmentation of the nucleus of a dying cell whereby its chromatin is distributed irregularly throughout the cytoplasm. It is usually preceded by pyknosis and can occur as a result of either programmed cell death (apoptosis), cellular senescence, or necrosis.
Acute promyelocytic leukemia is a subtype of acute myeloid leukemia (AML), a cancer of the white blood cells. In APL, there is an abnormal accumulation of immature granulocytes called promyelocytes. The disease is characterized by a chromosomal translocation involving the retinoic acid receptor alpha (RARA) gene and is distinguished from other forms of AML by its responsiveness to all-trans retinoic acid therapy. Acute promyelocytic leukemia was first characterized in 1957 by French and Norwegian physicians as a hyperacute fatal illness, with a median survival time of less than a week. Today, prognoses have drastically improved; 10-year survival rates are estimated to be approximately 80-90% according to one study.
Fas ligand is a type-II transmembrane protein expressed on various types of cells, including cytotoxic T lymphocytes, monocytes, neutrophils, breast epithelial cells, vascular endothelial cells and natural killer (NK) cells. It binds with its receptor, called FAS receptor and plays a crucial role in the regulation of the immune system and in induction of apoptosis, a programmed cell death.
Arsenic trioxide is an inorganic compound with the formula As 2O 3. As an industrial chemical, its major uses include the manufacture of wood preservatives, pesticides, and glass. It is sold under the brand name Trisenox among others when used as a medication to treat a type of cancer known as acute promyelocytic leukemia. For this use it is given by injection into a vein.
The apoptosome is a large quaternary protein structure formed in the process of apoptosis. Its formation is triggered by the release of cytochrome c from the mitochondria in response to an internal (intrinsic) or external (extrinsic) cell death stimulus. Stimuli can vary from DNA damage and viral infection to developmental cues such as those leading to the degradation of a tadpole's tail.
Apoptosis regulator BAX, also known as bcl-2-like protein 4, is a protein that in humans is encoded by the BAX gene. BAX is a member of the Bcl-2 gene family. BCL2 family members form hetero- or homodimers and act as anti- or pro-apoptotic regulators that are involved in a wide variety of cellular activities. This protein forms a heterodimer with BCL2, and functions as an apoptotic activator. This protein is reported to interact with, and increase the opening of, the mitochondrial voltage-dependent anion channel (VDAC), which leads to the loss in membrane potential and the release of cytochrome c. The expression of this gene is regulated by the tumor suppressor P53 and has been shown to be involved in P53-mediated apoptosis.
The BH3 interacting-domain death agonist, or BID, gene is a pro-apoptotic member of the Bcl-2 protein family. Bcl-2 family members share one or more of the four characteristic domains of homology entitled the Bcl-2 homology (BH) domains, and can form hetero- or homodimers. Bcl-2 proteins act as anti- or pro-apoptotic regulators that are involved in a wide variety of cellular activities.
The p53 upregulated modulator of apoptosis (PUMA) also known as Bcl-2-binding component 3 (BBC3), is a pro-apoptotic protein, member of the Bcl-2 protein family. In humans, the Bcl-2-binding component 3 protein is encoded by the BBC3 gene. The expression of PUMA is regulated by the tumor suppressor p53. PUMA is involved in p53-dependent and -independent apoptosis induced by a variety of signals, and is regulated by transcription factors, not by post-translational modifications. After activation, PUMA interacts with antiapoptotic Bcl-2 family members, thus freeing Bax and/or Bak which are then able to signal apoptosis to the mitochondria. Following mitochondrial dysfunction, the caspase cascade is activated ultimately leading to cell death.
Survivin, also called baculoviral inhibitor of apoptosis repeat-containing 5 or BIRC5, is a protein that, in humans, is encoded by the BIRC5 gene.
The HL-60 cell line is a human leukemia cell line that has been used for laboratory research on blood cell formation and physiology. HL-60 proliferates continuously in suspension culture in nutrient and antibiotic chemicals. The doubling time is about 36–48 hours. The cell line was derived from a 36-year-old woman who was originally reported to have acute promyelocytic leukemia at the MD Anderson Cancer Center. HL-60 cells predominantly show neutrophilic promyelocytic morphology. Subsequent evaluation, including the karyotype that showed absence of the defining t(15;17) translocation, concluded that HL-60 cells are from a case of AML FAB-M2.
B-cell lymphoma-extra large (Bcl-xL), encoded by the BCL2-like 1 gene, is a transmembrane molecule in the mitochondria. It is a member of the Bcl-2 family of proteins, and acts as an anti-apoptotic protein by preventing the release of mitochondrial contents such as cytochrome c, which leads to caspase activation and ultimately, programmed cell death.
Acute myeloblastic leukemia with maturation (M2) is a subtype of acute myeloid leukemia (AML).
Induced myeloid leukemia cell differentiation protein Mcl-1 is a protein that in humans is encoded by the MCL1 gene.
Diablo homolog (DIABLO) is a mitochondrial protein that in humans is encoded by the DIABLO gene on chromosome 12. DIABLO is also referred to as second mitochondria-derived activator of caspases or SMAC. This protein binds inhibitor of apoptosis proteins (IAPs), thus freeing caspases to activate apoptosis. Due to its proapoptotic function, SMAC is implicated in a broad spectrum of tumors, and small molecule SMAC mimetics have been developed to enhance current cancer treatments.
Gene expression profiling has revealed that diffuse large B-cell lymphoma (DLBCL) is composed of at least 3 different sub-groups, each having distinct oncogenic mechanisms that respond to therapies in different ways. Germinal Center B-Cell like (GCB) DLBCLs appear to arise from normal germinal center B cells, while Activated B-cell like (ABC) DLBCLs are thought to arise from postgerminal center B cells that are arrested during plasmacytic differentiation. The differences in gene expression between GCB DLBCL and ABC DLBCL are as vast as the differences between distinct types of leukemia, but these conditions have historically been grouped together and treated as the same disease.
Antineoplastic resistance, often used interchangeably with chemotherapy resistance, is the resistance of neoplastic (cancerous) cells, or the ability of cancer cells to survive and grow despite anti-cancer therapies. In some cases, cancers can evolve resistance to multiple drugs, called multiple drug resistance.
Paraptosis is a type of programmed cell death, morphologically distinct from apoptosis and necrosis. The defining features of paraptosis are cytoplasmic vacuolation, independent of caspase activation and inhibition, and lack of apoptotic morphology. Paraptosis lacks several of the hallmark characteristics of apoptosis, such as membrane blebbing, chromatin condensation, and nuclear fragmentation. Like apoptosis and other types of programmed cell death, the cell is involved in causing its own death, and gene expression is required. This is in contrast to necrosis, which is non-programmed cell death that results from injury to the cell.
References
1 2 Modrzejewski, Feliks (1977). Farmacja stosowana, podręcznik dla studentów farmacji[Applied Pharmacy: A Textbook for Pharmacy Students] (in Polish). Vol.V. Warsaw: Państwowy Zakład Wydawnictw Lekarskich. p.384.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.