Related Research Articles

Ehlers–Danlos syndromes (EDS) are a group of 13 genetic connective-tissue disorders in the current classification, with the latest type discovered in 2018. Symptoms include loose joints, joint pain, stretchy velvety skin, and abnormal scar formation. These may be noticed at birth or in early childhood. Complications may include aortic dissection, joint dislocations, scoliosis, chronic pain, or early osteoarthritis.
Hypotonia is a state of low muscle tone, often involving reduced muscle strength. Hypotonia is not a specific medical disorder, but a potential manifestation of many different diseases and disorders that affect motor nerve control by the brain or muscle strength. Hypotonia is a lack of resistance to passive movement, whereas muscle weakness results in impaired active movement. Central hypotonia originates from the central nervous system, while peripheral hypotonia is related to problems within the spinal cord, peripheral nerves and/or skeletal muscles. Severe hypotonia in infancy is commonly known as floppy baby syndrome. Recognizing hypotonia, even in early infancy, is usually relatively straightforward, but diagnosing the underlying cause can be difficult and often unsuccessful. The long-term effects of hypotonia on a child's development and later life depend primarily on the severity of the muscle weakness and the nature of the cause. Some disorders have a specific treatment but the principal treatment for most hypotonia of idiopathic or neurologic cause is physical therapy and/or occupational therapy for remediation.
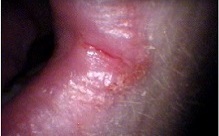
Plummer–Vinson syndrome is a rare disease characterized by difficulty swallowing, iron-deficiency anemia, glossitis, cheilosis and esophageal webs. Treatment with iron supplementation and mechanical widening of the esophagus generally provides an excellent outcome.

Olivopontocerebellar atrophy (OPCA) is the degeneration of neurons in specific areas of the brain – the cerebellum, pons, and inferior olivary nucleus. OPCA is present in several neurodegenerative syndromes, including inherited and non-inherited forms of ataxia and multiple system atrophy (MSA), with which it is primarily associated.

Crouzon syndrome is an autosomal dominant genetic disorder known as a branchial arch syndrome. Specifically, this syndrome affects the first branchial arch, which is the precursor of the maxilla and mandible. Since the branchial arches are important developmental features in a growing embryo, disturbances in their development create lasting and widespread effects.

Cockayne syndrome (CS), also called Neill-Dingwall syndrome, is a rare and fatal autosomal recessive neurodegenerative disorder characterized by growth failure, impaired development of the nervous system, abnormal sensitivity to sunlight (photosensitivity), eye disorders and premature aging. Failure to thrive and neurological disorders are criteria for diagnosis, while photosensitivity, hearing loss, eye abnormalities, and cavities are other very common features. Problems with any or all of the internal organs are possible. It is associated with a group of disorders called leukodystrophies, which are conditions characterized by degradation of neurological white matter. The underlying disorder is a defect in a DNA repair mechanism. Unlike other defects of DNA repair, patients with CS are not predisposed to cancer or infection. Cockayne syndrome is a rare but destructive disease usually resulting in death within the first or second decade of life. The mutation of specific genes in Cockayne syndrome is known, but the widespread effects and its relationship with DNA repair is yet to be well understood.
Trisomy 8 causes Warkany syndrome 2, a human chromosomal disorder caused by having three copies (trisomy) of chromosome 8. It can appear with or without mosaicism.

Carpenter syndrome, also called acrocephalopolysyndactyly type II, is an extremely rare autosomal recessive congenital disorder characterized by craniofacial malformations, obesity, syndactyly, and polydactyly. Acrocephalopolysyndactyly is a variation of acrocephalosyndactyly that presents with polydactyly.

Behr syndrome is characterized by the association of early-onset optic atrophy with spinocerebellar degeneration resulting in ataxia, pyramidal signs, peripheral neuropathy and developmental delay.

GAPO syndrome is a rare, autosomal recessive disorder that causes severe growth retardation, and has been observed fewer than 30 times before 2011. GAPO is an acronym that encompasses the predominant traits of the disorder: growth retardation, alopecia, pseudoanodontia, and worsening optic atrophy in some subjects. Other common symptoms include premature aging, large, prominent foreheads, and delayed bone aging. GAPO syndrome typically results in premature death around age 30–40, due to interstitial fibrosis and atherosclerosis.

Greig cephalopolysyndactyly syndrome is a disorder that affects development of the limbs, head, and face. The features of this syndrome are highly variable, ranging from very mild to severe. People with this condition typically have one or more extra fingers or toes (polydactyly) or an abnormally wide thumb or big toe (hallux).

Duane-radial ray syndrome, also known as Okihiro Syndrome, is a rare autosomal dominant disorder that primarily affects the eyes and causes abnormalities of bones in the arms and hands. This disorder is considered to be a SALL4-related disorder due to the SALL4 gene mutations leading to these abnormalities. It is diagnosed by clinical findings on a physical exam as well as genetic testing and imaging. After being diagnosed, there are other evaluations that one may go through in order to determine the extent of the disease. There are various treatments for the symptoms of this disorder.

Papillorenal syndrome is an autosomal dominant genetic disorder marked by underdevelopment (hypoplasia) of the kidney and colobomas of the optic nerve.
Rabson–Mendenhall syndrome is a rare autosomal recessive disorder characterized by severe insulin resistance. The disorder is caused by mutations in the insulin receptor gene. Symptoms include growth abnormalities of the head, face and nails, along with the development of acanthosis nigricans. Treatment involves controlling blood glucose levels by using insulin and incorporating a strategically planned, controlled diet. Also, direct actions against other symptoms may be taken This syndrome usually affects children and has a prognosis of 1–2 years.
Antley–Bixler syndrome is a rare, severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.
3-M syndrome or 3M3 is a rare hereditary disorder characterized by severe growth retardation, facial dysmorphia, and skeletal abnormalities. The name 3-M is derived from the initials of the three researchers who first identified it: Miller, McKusick, and Malvaux and report their findings in the medical literature in 1972. Mutations in any one of the following three genes: CUL7, OBSL1, and CCDC8 are responsible for the occurrence of this disorder. It is inherited through an autosomal recessive pattern and considered very rare, so far less than 100 cases worldwide have been identified. Diagnosis is based on the presence of clinical features. Genetic testing can confirm the diagnosis and identify the specific gene involved. Treatment is aimed at addressing the growth and skeletal problems and may include surgical bone lengthening, adaptive aids, and physical therapy. An endocrinologist may assist with growth hormone replacement and appropriate evaluations during puberty.
Marinesco–Sjögren syndrome (MSS), sometimes spelled Marinescu–Sjögren syndrome, is a rare autosomal recessive disorder.
Gillespie syndrome, also called aniridia, cerebellar ataxia and mental deficiency, is a rare genetic disorder. The disorder is characterized by partial aniridia, ataxia, and, in most cases, intellectual disability. It is heterogeneous, inherited in either an autosomal dominant or autosomal recessive manner. Gillespie syndrome was first described by American ophthalmologist Fredrick Gillespie in 1965.
Donohue syndrome is an extremely rare and severe genetic disorder. Leprechaunism derives its name from the hallmark elvish features exhibited by the affected individuals. The disease is caused by a mutation in the INSR gene, which contains the genetic information for the formation of insulin receptors. As a result, affected individuals have either a decreased number of insulin receptors, or insulin receptor with greatly impaired functionality. The lack and impairment of insulin receptor functionality leads to an inability to regulate blood glucose levels through severe insulin resistance. This will ultimately lead to affected development of tissues and organs throughout the body. In addition to the physical abnormalities, leprechaunism is also characterized by endocrine system abnormalities that can lead to conditions such as hyperglycemia, hypoglycemia, hyperinsulemia, and the enlargement of certain sex organs such as the penis in males, and the clitoris in females.
Baller–Gerold syndrome (BGS) is a rare genetic syndrome that involves premature fusion of the skull bones and malformations of facial, forearm and hand bones. The symptoms of Baller–Gerold syndrome overlap with features of a few other genetics disorders: Rothmund–Thomson syndrome and RAPADILINO syndrome. The prevalence of BGS is unknown, as there have only been a few reported cases, but it is estimated to be less than 1 in a million. The name of the syndrome comes from the researchers Baller and Gerold who discovered the first three cases.
References
- ↑ "Definition of Microorchidism". MedicineNet. Retrieved 2009-08-03.
| | This genetic disorder article is a stub. You can help Wikipedia by expanding it. |