
MOLPRO is a software package used for accurate ab initio quantum chemistry calculations. It is developed by Peter Knowles at Cardiff University and Hans-Joachim Werner at Universität Stuttgart in collaboration with other authors.
Gaussian is a general purpose computational chemistry software package initially released in 1970 by John Pople and his research group at Carnegie Mellon University as Gaussian 70. It has been continuously updated since then. The name originates from Pople's use of Gaussian orbitals to speed up molecular electronic structure calculations as opposed to using Slater-type orbitals, a choice made to improve performance on the limited computing capacities of then-current computer hardware for Hartree–Fock calculations. The current version of the program is Gaussian 16. Originally available through the Quantum Chemistry Program Exchange, it was later licensed out of Carnegie Mellon University, and since 1987 has been developed and licensed by Gaussian, Inc.
Q-Chem is a general-purpose electronic structure package featuring a variety of established and new methods implemented using innovative algorithms that enable fast calculations of large systems on various computer architectures, from laptops and regular lab workstations to midsize clusters, HPCC, and cloud computing using density functional and wave-function based approaches. It offers an integrated graphical interface and input generator; a large selection of functionals and correlation methods, including methods for electronically excited states and open-shell systems; solvation models; and wave-function analysis tools. In addition to serving the computational chemistry community, Q-Chem also provides a versatile code development platform.
Psi is an ab initio computational chemistry package originally written by the research group of Henry F. Schaefer, III. Utilizing Psi, one can perform a calculation on a molecular system with various kinds of methods such as Hartree-Fock, Post-Hartree–Fock electron correlation methods, and density functional theory. The program can compute energies, optimize molecular geometries, and compute vibrational frequencies. The major part of the program is written in C++, while Python API is also available, which allows users to perform complex computations or automate tasks easily.
Electronic correlation is the interaction between electrons in the electronic structure of a quantum system. The correlation energy is a measure of how much the movement of one electron is influenced by the presence of all other electrons.
In computational chemistry, post–Hartree–Fock (post-HF) methods are the set of methods developed to improve on the Hartree–Fock (HF), or self-consistent field (SCF) method. They add electron correlation which is a more accurate way of including the repulsions between electrons than in the Hartree–Fock method where repulsions are only averaged.

MOLCAS is an ab initio computational chemistry program, developed as a joint project by a number of international institutes. MOLCAS is developed by scientists to be used by scientists. It is not primarily a commercial product and it is not sold in order to produce a fortune for its owner.
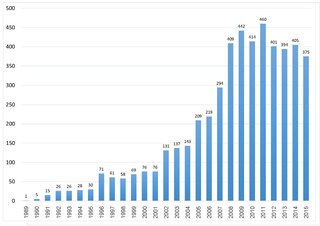
TURBOMOLE is an ab initio computational chemistry program that implements various quantum chemistry methods. It was initially developed by the group of Prof. Reinhart Ahlrichs at the University of Karlsruhe. In 2007, TURBOMOLE GmbH, founded by R. Ahlrichs, F. Furche, C. Hättig, W. Klopper, M. Sierka, and F. Weigend, took over the responsibility for the coordination of the scientific development of TURBOMOLE program, for which the company holds all copy and intellectual property rights. In 2018 David P. Tew joined the TURBOMOLE GmbH. Since 1987, this program is one of the useful tools as it involves in many fields of research including heterogeneous and homogeneous catalysis, organic and inorganic chemistry, spectroscopy as well as biochemistry. This can be illustrated by citation records of Ahlrich's 1989 publication which is more than 6700 times as of 18 July 2020. In the year 2014, the second Turbomole article has been published. The number of citations from both papers indicates that the Turbomole's user base is expanding.
CRYSTAL is a quantum chemistry ab initio program, designed primarily for calculations on crystals, slabs and polymers using translational symmetry, but it can also be used for single molecules. It is written by V.R. Saunders, R. Dovesi, C. Roetti, R. Orlando, C.M. Zicovich-Wilson, N.M. Harrison, K. Doll, B. Civalleri, I.J. Bush, Ph. D’Arco, and M. Llunell from Theoretical Chemistry Group at the University of Torino and the Computational Materials Science Group at the Daresbury Laboratory near Warrington in Cheshire, England. The current version is CRYSTAL23. Earlier versions were CRYSTAL88, CRYSTAL92, CRYSTAL95, CRYSTAL98, CRYSTAL03, CRYSTAL06, CRYSTAL09, CRYSTAL14, and CRYSTAL17.
Jaguar is a computer software package used for ab initio quantum chemistry calculations for both gas and solution phases. It is commercial software marketed by the company Schrödinger. The program was originated in research groups of Richard Friesner and William Goddard and was initially called PS-GVB.
The COLUMBUS PROGRAMS are a computational chemistry software suite for calculating ab initio molecular electronic structures, designed as a collection of individual programs communicating through files. The programs focus on extended multi-reference calculations of atomic and molecular ground and excited states. In addition to standard classes of reference wave functions such as CAS and RAS, calculations can be performed with selected configurations. Some features employ the atomic orbital integrals and gradient routines from the Dalton as well as MOLCAS program suites. COLUMBUS is distributed open-source under the LGPL license.
General Atomic and Molecular Electronic Structure System (GAMESS-UK) is a computer software program for computational chemistry. The original code split in 1981 into GAMESS-UK and GAMESS (US) variants, which now differ significantly. Many of the early developments in the UK version arose from the earlier UK based ATMOL program, which, unlike GAMESS, lacked analytical gradients for geometry optimisation.

Spartan is a molecular modelling and computational chemistry application from Wavefunction. It contains code for molecular mechanics, semi-empirical methods, ab initio models, density functional models, post-Hartree–Fock models, and thermochemical recipes including G3(MP2) and T1. Quantum chemistry calculations in Spartan are powered by Q-Chem.
Ab initio quantum chemistry methods are computational chemistry methods based on quantum chemistry. The term ab initio was first used in quantum chemistry by Robert Parr and coworkers, including David Craig in a semiempirical study on the excited states of benzene. The background is described by Parr. Ab initio means "from first principles" or "from the beginning", implying that the only inputs into an ab initio calculation are physical constants. Ab initio quantum chemistry methods attempt to solve the electronic Schrödinger equation given the positions of the nuclei and the number of electrons in order to yield useful information such as electron densities, energies and other properties of the system. The ability to run these calculations has enabled theoretical chemists to solve a range of problems and their importance is highlighted by the awarding of the Nobel prize to John Pople and Walter Kohn.
Quantum chemistry composite methods are computational chemistry methods that aim for high accuracy by combining the results of several calculations. They combine methods with a high level of theory and a small basis set with methods that employ lower levels of theory with larger basis sets. They are commonly used to calculate thermodynamic quantities such as enthalpies of formation, atomization energies, ionization energies and electron affinities. They aim for chemical accuracy which is usually defined as within 1 kcal/mol of the experimental value. The first systematic model chemistry of this type with broad applicability was called Gaussian-1 (G1) introduced by John Pople. This was quickly replaced by the Gaussian-2 (G2) which has been used extensively. The Gaussian-3 (G3) was introduced later.
In the field of computational chemistry, energy minimization is the process of finding an arrangement in space of a collection of atoms where, according to some computational model of chemical bonding, the net inter-atomic force on each atom is acceptably close to zero and the position on the potential energy surface (PES) is a stationary point. The collection of atoms might be a single molecule, an ion, a condensed phase, a transition state or even a collection of any of these. The computational model of chemical bonding might, for example, be quantum mechanics.

CP2K is a freely available (GPL) quantum chemistry and solid state physics program package, written in Fortran 2008, to perform atomistic simulations of solid state, liquid, molecular, periodic, material, crystal, and biological systems. It provides a general framework for different methods: density functional theory (DFT) using a mixed Gaussian and plane waves approach (GPW) via LDA, GGA, MP2, or RPA levels of theory, classical pair and many-body potentials, semi-empirical and tight-binding Hamiltonians, as well as Quantum Mechanics/Molecular Mechanics (QM/MM) hybrid schemes relying on the Gaussian Expansion of the Electrostatic Potential (GEEP). The Gaussian and Augmented Plane Waves method (GAPW) as an extension of the GPW method allows for all-electron calculations. CP2K can do simulations of molecular dynamics, metadynamics, Monte Carlo, Ehrenfest dynamics, vibrational analysis, core level spectroscopy, energy minimization, and transition state optimization using NEB or dimer method.
The ONIOM method is a computational approach developed by Keiji Morokuma and co-workers. ONIOM is a hybrid method that enables different ab initio, semi-empirical, or molecular mechanics methods to be applied to different parts of a molecule/system in combination to produce reliable geometry and energy at reduced computational cost.
TeraChem is a computational chemistry software program designed for CUDA-enabled Nvidia GPUs. The initial development started at the University of Illinois at Urbana-Champaign and was subsequently commercialized. It is currently distributed by PetaChem, LLC, located in Silicon Valley. As of 2020, the software package is still under active development.
Python-based Simulations of Chemistry Framework (PySCF) is an ab initio computational chemistry program natively implemented in Python program language. The package aims to provide a simple, light-weight and efficient platform for quantum chemistry code developing and calculation. It provides various functions to do the Hartree–Fock, MP2, density functional theory, MCSCF, coupled cluster theory at non-relativistic level and 4-component relativistic Hartree–Fock theory. Although most functions are written in Python, the computation critical modules are intensively optimized in C. As a result, the package works as efficient as other C/Fortran-based quantum chemistry program. PySCF is developed by Qiming Sun. PySCF2.0 is the latest version of the program.
