Related Research Articles

Valence shell electron pair repulsion (VSEPR) theory, is a model used in chemistry to predict the geometry of individual molecules from the number of electron pairs surrounding their central atoms. It is also named the Gillespie-Nyholm theory after its two main developers, Ronald Gillespie and Ronald Nyholm.
The extended Hückel method is a semiempirical quantum chemistry method, developed by Roald Hoffmann since 1963. It is based on the Hückel method but, while the original Hückel method only considers pi orbitals, the extended method also includes the sigma orbitals.
The Vienna Ab initio Simulation Package, better known as VASP, is a package for performing ab initio quantum mechanical calculations using either Vanderbilt pseudopotentials, or the projector augmented wave method, and a plane wave basis set. The basic methodology is density functional theory (DFT), but the code also allows use of post-DFT corrections such as hybrid functionals mixing DFT and Hartree–Fock exchange, many-body perturbation theory and dynamical electronic correlations within the random phase approximation (RPA) and MP2.
The Jahn–Teller effect is an important mechanism of spontaneous symmetry breaking in molecular and solid-state systems which has far-reaching consequences in different fields, and is responsible for a variety of phenomena in spectroscopy, stereochemistry, crystal chemistry, molecular and solid-state physics, and materials science. The effect is named for Hermann Arthur Jahn and Edward Teller, who first reported studies about it in 1937. The Jahn–Teller effect, and the related Renner–Teller effect, are discussed in Section 13.4 of the spectroscopy textbook by Bunker and Jensen.
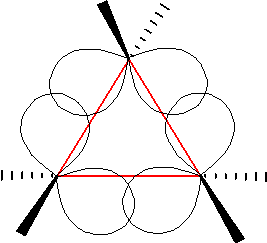
In organic chemistry, a bent bond, also known as a banana bond, is a type of covalent chemical bond with a geometry somewhat reminiscent of a banana. The term itself is a general representation of electron density or configuration resembling a similar "bent" structure within small ring molecules, such as cyclopropane (C3H6) or as a representation of double or triple bonds within a compound that is an alternative to the sigma and pi bond model.

In classical mechanics, anharmonicity is the deviation of a system from being a harmonic oscillator. An oscillator that is not oscillating in harmonic motion is known as an anharmonic oscillator where the system can be approximated to a harmonic oscillator and the anharmonicity can be calculated using perturbation theory. If the anharmonicity is large, then other numerical techniques have to be used. In reality all oscillating systems are anharmonic, but most approximate the harmonic oscillator the smaller the amplitude of the oscillation is.

Spartan is a molecular modelling and computational chemistry application from Wavefunction. It contains code for molecular mechanics, semi-empirical methods, ab initio models, density functional models, post-Hartree–Fock models, and thermochemical recipes including G3(MP2) and T1. Quantum chemistry calculations in Spartan are powered by Q-Chem.
Uranocene, U(C8H8)2, is an organouranium compound composed of a uranium atom sandwiched between two cyclooctatetraenide rings. It was one of the first organoactinide compounds to be synthesized. It is a green air-sensitive solid that dissolves in organic solvents. Uranocene, a member of the "actinocenes," a group of metallocenes incorporating elements from the actinide series. It is the most studied bis[8]annulene-metal system, although it has no known practical applications.

Carbon trioxide (CO3) is an unstable oxide of carbon (an oxocarbon). The possible isomers of carbon trioxide include ones with molecular symmetry point groups Cs, D3h, and C2v. The C2v state, consisting of a dioxirane, has been shown to be the ground state of the molecule. Carbon trioxide should not be confused with the stable carbonate ion (CO2−
3).

In computational chemistry, a water model is used to simulate and thermodynamically calculate water clusters, liquid water, and aqueous solutions with explicit solvent. The models are determined from quantum mechanics, molecular mechanics, experimental results, and these combinations. To imitate a specific nature of molecules, many types of models have been developed. In general, these can be classified by the following three points; (i) the number of interaction points called site, (ii) whether the model is rigid or flexible, (iii) whether the model includes polarization effects.
This page deals with the electron affinity as a property of isolated atoms or molecules. Solid state electron affinities are not listed here.
Hybrid functionals are a class of approximations to the exchange–correlation energy functional in density functional theory (DFT) that incorporate a portion of exact exchange from Hartree–Fock theory with the rest of the exchange–correlation energy from other sources. The exact exchange energy functional is expressed in terms of the Kohn–Sham orbitals rather than the density, so is termed an implicit density functional. One of the most commonly used versions is B3LYP, which stands for "Becke, 3-parameter, Lee–Yang–Parr".
PARSEC is a package designed to perform electronic structure calculations of solids and molecules using density functional theory (DFT). The acronym stands for Pseudopotential Algorithm for Real-Space Electronic Calculations. It solves the Kohn–Sham equations in real space, without the use of explicit basis sets.
Photofragment ion imaging or, more generally, Product Imaging is an experimental technique for making measurements of the velocity of product molecules or particles following a chemical reaction or the photodissociation of a parent molecule. The method uses a two-dimensional detector, usually a microchannel plate, to record the arrival positions of state-selected ions created by resonantly enhanced multi-photon ionization (REMPI). The first experiment using photofragment ion imaging was performed by David W Chandler and Paul L Houston in 1987 on the phototodissociation dynamics of methyl iodide (iodomethane, CH3I).
MADNESS is a high-level software environment for the solution of integral and differential equations in many dimensions using adaptive and fast harmonic analysis methods with guaranteed precision based on multiresolution analysis and separated representations .
BigDFT is a free software package for physicists and chemists, distributed under the GNU General Public License, whose main program allows the total energy, charge density, and electronic structure of systems made of electrons and nuclei to be calculated within density functional theory (DFT), using pseudopotentials, and a wavelet basis.
Xenon monochloride (XeCl) is an exciplex which is used in excimer lasers and excimer lamps emitting near ultraviolet light at 308 nm. It is most commonly used in medicine. Xenon monochloride was first synthesized in the 1960s. Its kinetic scheme is very complex and its state changes occur on a nanosecond timescale. In the gaseous state, at least two kinds of xenon monochloride are known: XeCl and Xe
2Cl, whereas complex aggregates form in the solid state in noble gas matrices. The excited state of xenon resembles halogens and it reacts with them to form excited molecular compounds.

James Bernhard Anderson was an American chemist and physicist. From 1995 to 2014 he was Evan Pugh Professor of Chemistry and Physics at the Pennsylvania State University. He specialized in Quantum Chemistry by Monte Carlo methods, molecular dynamics of reactive collisions, kinetics and mechanisms of gas phase reactions, and rare-event theory.

Bidyendu Mohan Deb is an Indian theoretical chemist, chemical physicist and a professor at the Indian Institute of Science Education and Research, Kolkata (IISER). he is known for his studies in theoretical chemistry and chemical physics. He is an elected fellow of the International Union of Pure and Applied Chemistry, The World Academy of Sciences, Indian National Science Academy and the Indian Academy of Sciences. The Council of Scientific and Industrial Research, the apex agency of the Government of India for scientific research, awarded him the Shanti Swarup Bhatnagar Prize for Science and Technology, one of the highest Indian science awards, in 1981, for his contributions to chemical sciences.
FHI-aims is a software package for computational molecular and materials science written in Fortran. It uses density functional theory and many-body perturbation theory to simulate chemical and physical properties of atoms, molecules, nanostructures, solids, and surfaces. Originally developed at the Fritz Haber Institute in Berlin, the ongoing development of the FHI-aims source code is now driven by a worldwide community of collaborating research institutions.
References
- ↑ B. Delley (1990). "An All-Electron Numerical Method for Solving the Local Density Functional for Polyatomic Molecules". J. Chem. Phys. 92 (1): 508–517. Bibcode:1990JChPh..92..508D. doi:10.1063/1.458452.
- ↑ B. Delley (2000). "From molecules to solids with the DMol3 approach". J. Chem. Phys. 113 (18): 7756–7764. Bibcode:2000JChPh.113.7756D. doi:10.1063/1.1316015.
- ↑ J. Andzelm C. Kölmel A. Klamt (1995). "Incorporation of solvent effects into density-functional calculations of molecular energies and geometries". J. Chem. Phys. 103 (21): 9312–9320. Bibcode:1995JChPh.103.9312A. doi:10.1063/1.469990.
- ↑ B. Delley D. Ellis A. Freeman E. Baerends D. Post (1983). "Binding Energy and Electronic Structure of Small Copper Particles". Phys. Rev. B. 27 (4): 2132–2144. Bibcode:1983PhRvB..27.2132D. doi:10.1103/PhysRevB.27.2132. hdl: 1871/10013 .
- ↑ "Citations for An all-electron numerical method for solving the local density functional for polyatomic molecules".
| | This quantum chemistry-related article is a stub. You can help Wikipedia by expanding it. |