
Computational chemistry is a branch of chemistry that uses computer simulations to assist in solving chemical problems. It uses methods of theoretical chemistry incorporated into computer programs to calculate the structures and properties of molecules, groups of molecules, and solids. The importance of this subject stems from the fact that, with the exception of some relatively recent findings related to the hydrogen molecular ion, achieving an accurate quantum mechanical depiction of chemical systems analytically, or in a closed form, is not feasible. The complexity inherent in the many-body problem exacerbates the challenge of providing detailed descriptions of quantum mechanical systems. While computational results normally complement information obtained by chemical experiments, it can occasionally predict unobserved chemical phenomena.

Molecular dynamics (MD) is a computer simulation method for analyzing the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period of time, giving a view of the dynamic "evolution" of the system. In the most common version, the trajectories of atoms and molecules are determined by numerically solving Newton's equations of motion for a system of interacting particles, where forces between the particles and their potential energies are often calculated using interatomic potentials or molecular mechanical force fields. The method is applied mostly in chemical physics, materials science, and biophysics.
Ewald summation, named after Paul Peter Ewald, is a method for computing long-range interactions in periodic systems. It was first developed as the method for calculating the electrostatic energies of ionic crystals, and is now commonly used for calculating long-range interactions in computational chemistry. Ewald summation is a special case of the Poisson summation formula, replacing the summation of interaction energies in real space with an equivalent summation in Fourier space. In this method, the long-range interaction is divided into two parts: a short-range contribution, and a long-range contribution which does not have a singularity. The short-range contribution is calculated in real space, whereas the long-range contribution is calculated using a Fourier transform. The advantage of this method is the rapid convergence of the energy compared with that of a direct summation. This means that the method has high accuracy and reasonable speed when computing long-range interactions, and it is thus the de facto standard method for calculating long-range interactions in periodic systems. The method requires charge neutrality of the molecular system to accurately calculate the total Coulombic interaction. A study of the truncation errors introduced in the energy and force calculations of disordered point-charge systems is provided by Kolafa and Perram.
Car–Parrinello molecular dynamics or CPMD refers to either a method used in molecular dynamics or the computational chemistry software package used to implement this method.

In computational chemistry, a water model is used to simulate and thermodynamically calculate water clusters, liquid water, and aqueous solutions with explicit solvent. The models are determined from quantum mechanics, molecular mechanics, experimental results, and these combinations. To imitate a specific nature of molecules, many types of models have been developed. In general, these can be classified by the following three points; (i) the number of interaction points called site, (ii) whether the model is rigid or flexible, (iii) whether the model includes polarization effects.
In computational chemistry, a constraint algorithm is a method for satisfying the Newtonian motion of a rigid body which consists of mass points. A restraint algorithm is used to ensure that the distance between mass points is maintained. The general steps involved are: (i) choose novel unconstrained coordinates, (ii) introduce explicit constraint forces, (iii) minimize constraint forces implicitly by the technique of Lagrange multipliers or projection methods.
In computer simulations of mechanical systems, energy drift is the gradual change in the total energy of a closed system over time. According to the laws of mechanics, the energy should be a constant of motion and should not change. However, in simulations the energy might fluctuate on a short time scale and increase or decrease on a very long time scale due to numerical integration artifacts that arise with the use of a finite time step Δt. This is somewhat similar to the flying ice cube problem, whereby numerical errors in handling equipartition of energy can change vibrational energy into translational energy.

CP2K is a freely available (GPL) quantum chemistry and solid state physics program package, written in Fortran 2008, to perform atomistic simulations of solid state, liquid, molecular, periodic, material, crystal, and biological systems. It provides a general framework for different methods: density functional theory (DFT) using a mixed Gaussian and plane waves approach (GPW) via LDA, GGA, MP2, or RPA levels of theory, classical pair and many-body potentials, semi-empirical and tight-binding Hamiltonians, as well as Quantum Mechanics/Molecular Mechanics (QM/MM) hybrid schemes relying on the Gaussian Expansion of the Electrostatic Potential (GEEP). The Gaussian and Augmented Plane Waves method (GAPW) as an extension of the GPW method allows for all-electron calculations. CP2K can do simulations of molecular dynamics, metadynamics, Monte Carlo, Ehrenfest dynamics, vibrational analysis, core level spectroscopy, energy minimization, and transition state optimization using NEB or dimer method.
D. E. Shaw Research (DESRES) is a privately held biochemistry research company based in New York City. Under the scientific direction of David E. Shaw, the group's chief scientist, D. E. Shaw Research develops technologies for molecular dynamics simulations and applies such simulations to basic scientific research in structural biology and biochemistry, and to the process of computer-aided drug design.

Anton is a massively parallel supercomputer designed and built by D. E. Shaw Research in New York, first running in 2008. It is a special-purpose system for molecular dynamics (MD) simulations of proteins and other biological macromolecules. An Anton machine consists of a substantial number of application-specific integrated circuits (ASICs), interconnected by a specialized high-speed, three-dimensional torus network.
Particle–Particle–Particle–Mesh (P3M) is a Fourier-based Ewald summation method to calculate potentials in N-body simulations.
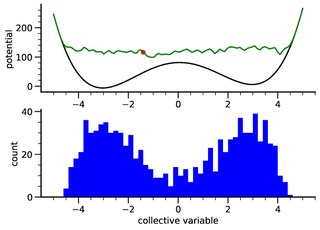
Metadynamics is a computer simulation method in computational physics, chemistry and biology. It is used to estimate the free energy and other state functions of a system, where ergodicity is hindered by the form of the system's energy landscape. It was first suggested by Alessandro Laio and Michele Parrinello in 2002 and is usually applied within molecular dynamics simulations. MTD closely resembles a number of newer methods such as adaptively biased molecular dynamics, adaptive reaction coordinate forces and local elevation umbrella sampling. More recently, both the original and well-tempered metadynamics were derived in the context of importance sampling and shown to be a special case of the adaptive biasing potential setting. MTD is related to the Wang–Landau sampling.
Local elevation is a technique used in computational chemistry or physics, mainly in the field of molecular simulation. It was developed in 1994 by Huber, Torda and van Gunsteren to enhance the searching of conformational space in molecular dynamics simulations and is available in the GROMOS software for molecular dynamics simulation. The method was, together with the conformational flooding method, the first to introduce memory dependence into molecular simulations. Many recent methods build on the principles of the local elevation technique, including the Engkvist-Karlström, adaptive biasing force, Wang–Landau, metadynamics, adaptively biased molecular dynamics, adaptive reaction coordinate forces, and local elevation umbrella sampling methods. The basic principle of the method is to add a memory-dependent potential energy term in the simulation so as to prevent the simulation to revisit already sampled configurations, which leads to the increased probability of discovering new configurations. The method can be seen as a continuous variant of the Tabu search method.
Path integral molecular dynamics (PIMD) is a method of incorporating quantum mechanics into molecular dynamics simulations using Feynman path integrals. In PIMD, one uses the Born–Oppenheimer approximation to separate the wavefunction into a nuclear part and an electronic part. The nuclei are treated quantum mechanically by mapping each quantum nucleus onto a classical system of several fictitious particles connected by springs governed by an effective Hamiltonian, which is derived from Feynman's path integral. The resulting classical system, although complex, can be solved relatively quickly. There are now a number of commonly used condensed matter computer simulation techniques that make use of the path integral formulation including Centroid Molecular Dynamics (CMD), Ring Polymer Molecular Dynamics (RPMD), and the Feynman-Kleinert Quasi-Classical Wigner (FK-QCW) method. The same techniques are also used in path integral Monte Carlo (PIMC).
Adaptive sampling is a technique used in computational molecular biology to efficiently simulate protein folding when coupled with molecular dynamics simulations.
Surface hopping is a mixed quantum-classical technique that incorporates quantum mechanical effects into molecular dynamics simulations. Traditional molecular dynamics assume the Born-Oppenheimer approximation, where the lighter electrons adjust instantaneously to the motion of the nuclei. Though the Born-Oppenheimer approximation is applicable to a wide range of problems, there are several applications, such as photoexcited dynamics, electron transfer, and surface chemistry where this approximation falls apart. Surface hopping partially incorporates the non-adiabatic effects by including excited adiabatic surfaces in the calculations, and allowing for 'hops' between these surfaces, subject to certain criteria.

Ali Alavi FRS is a professor of theoretical chemistry in the Department of Chemistry at the University of Cambridge and a Director of the Max Planck Institute for Solid State Research in Stuttgart.
Na+/H+ antiporter A (NhaA) family (TC# 2.A.33) contains a number of bacterial sodium-proton antiporter (SPAP) proteins. These are integral membrane proteins that catalyse the exchange of H+ for Na+ in a manner that is highly pH dependent. Homologues have been sequenced from a number of bacteria and archaea. Prokaryotes possess multiple paralogues. A representative list of the proteins that belong to the NhaA family can be found in the Transporter Classification Database.
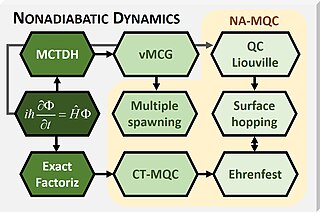
Mixed quantum-classical (MQC) dynamics is a class of computational theoretical chemistry methods tailored to simulate non-adiabatic (NA) processes in molecular and supramolecular chemistry. Such methods are characterized by:
- Propagation of nuclear dynamics through classical trajectories;
- Propagation of the electrons through quantum methods;
- A feedback algorithm between the electronic and nuclear subsystems to recover nonadiabatic information.