
Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.
A kinemage is an interactive graphic scientific illustration. It often is used to visualize molecules, especially proteins although it can also represent other types of 3-dimensional data. The kinemage system is designed to optimize ease of use, interactive performance, and the perception and communication of detailed 3D information. The kinemage information is stored in a text file, human- and machine-readable, that describes the hierarchy of display objects and their properties, and includes optional explanatory text. The kinemage format is a defined chemical MIME type of 'chemical/x-kinemage' with the file extension '.kin'.
Visual Molecular Dynamics (VMD) is a molecular modelling and visualization computer program. VMD is developed mainly as a tool to view and analyze the results of molecular dynamics simulations. It also includes tools for working with volumetric data, sequence data, and arbitrary graphics objects. Molecular scenes can be exported to external rendering tools such as POV-Ray, RenderMan, Tachyon, Virtual Reality Modeling Language (VRML), and many others. Users can run their own Tcl and Python scripts within VMD as it includes embedded Tcl and Python interpreters. VMD runs on Unix, Apple Mac macOS, and Microsoft Windows. VMD is available to non-commercial users under a distribution-specific license which permits both use of the program and modification of its source code, at no charge.
RasMol is a computer program written for molecular graphics visualization intended and used mainly to depict and explore biological macromolecule structures, such as those found in the Protein Data Bank. It was originally developed by Roger Sayle in the early 1990s.
Macromolecular docking is the computational modelling of the quaternary structure of complexes formed by two or more interacting biological macromolecules. Protein–protein complexes are the most commonly attempted targets of such modelling, followed by protein–nucleic acid complexes.

Molecular imaging is a field of medical imaging that focuses on imaging molecules of medical interest within living patients. This is in contrast to conventional methods for obtaining molecular information from preserved tissue samples, such as histology. Molecules of interest may be either ones produced naturally by the body, or synthetic molecules produced in a laboratory and injected into a patient by a doctor. The most common example of molecular imaging used clinically today is to inject a contrast agent into a patient's bloodstream and to use an imaging modality to track its movement in the body. Molecular imaging originated from the field of radiology from a need to better understand fundamental molecular processes inside organisms in a noninvasive manner.

Jmol is computer software for molecular modelling chemical structures in 3-dimensions. Jmol returns a 3D representation of a molecule that may be used as a teaching tool, or for research e.g., in chemistry and biochemistry. It is written in the programming language Java, so it can run on the operating systems Windows, macOS, Linux, and Unix, if Java is installed. It is free and open-source software released under a GNU Lesser General Public License (LGPL) version 2.0. A standalone application and a software development kit (SDK) exist that can be integrated into other Java applications, such as Bioclipse and Taverna.
In computational biology, protein pKa calculations are used to estimate the pKa values of amino acids as they exist within proteins. These calculations complement the pKa values reported for amino acids in their free state, and are used frequently within the fields of molecular modeling, structural bioinformatics, and computational biology.
A polyproline helix is a type of protein secondary structure which occurs in proteins comprising repeating proline residues. A left-handed polyproline II helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have trans isomers of their peptide bonds. This PPII conformation is also common in proteins and polypeptides with other amino acids apart from proline. Similarly, a more compact right-handed polyproline I helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have cis isomers of their peptide bonds. Of the twenty common naturally occurring amino acids, only proline is likely to adopt the cis isomer of the peptide bond, specifically the X-Pro peptide bond; steric and electronic factors heavily favor the trans isomer in most other peptide bonds. However, peptide bonds that replace proline with another N-substituted amino acid are also likely to adopt the cis isomer.

Sir Thomas Leon Blundell, is a British biochemist, structural biologist, and science administrator. He was a member of the team of Dorothy Hodgkin that solved in 1969 the first structure of a protein hormone, insulin. Blundell has made contributions to the structural biology of polypeptide hormones, growth factors, receptor activation, signal transduction, and DNA double-strand break repair, subjects important in cancer, tuberculosis, and familial diseases. He has developed software for protein modelling and understanding the effects of mutations on protein function, leading to new approaches to structure-guided and Fragment-based lead discovery. In 1999 he co-founded the oncology company Astex Therapeutics, which has moved ten drugs into clinical trials. Blundell has played central roles in restructuring British research councils and, as President of the UK Science Council, in developing professionalism in the practice of science.
Molecular biophysics is a rapidly evolving interdisciplinary area of research that combines concepts in physics, chemistry, engineering, mathematics and biology. It seeks to understand biomolecular systems and explain biological function in terms of molecular structure, structural organization, and dynamic behaviour at various levels of complexity. This discipline covers topics such as the measurement of molecular forces, molecular associations, allosteric interactions, Brownian motion, and cable theory. Additional areas of study can be found on Outline of Biophysics. The discipline has required development of specialized equipment and procedures capable of imaging and manipulating minute living structures, as well as novel experimental approaches.
The Database of Macromolecular Motions is a bioinformatics database and software-as-a-service tool that attempts to categorize macromolecular motions, sometimes also known as conformational change. It was originally developed by Mark B. Gerstein, Werner Krebs, and Nat Echols in the Molecular Biophysics & Biochemistry Department at Yale University.

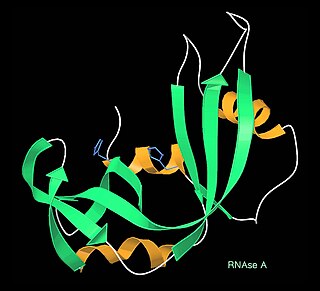
Ribbon diagrams, also known as Richardson diagrams, are 3D schematic representations of protein structure and are one of the most common methods of protein depiction used today. The ribbon depicts the general course and organisation of the protein backbone in 3D and serves as a visual framework for hanging details of the entire atomic structure, such as the balls for the oxygen atoms attached to myoglobin's active site in the adjacent figure. Ribbon diagrams are generated by interpolating a smooth curve through the polypeptide backbone. α-helices are shown as coiled ribbons or thick tubes, β-strands as arrows, and non-repetitive coils or loops as lines or thin tubes. The direction of the polypeptide chain is shown locally by the arrows, and may be indicated overall by a colour ramp along the length of the ribbon.
Phyre and Phyre2 are free web-based services for protein structure prediction. Phyre is among the most popular methods for protein structure prediction having been cited over 1500 times. Like other remote homology recognition techniques, it is able to regularly generate reliable protein models when other widely used methods such as PSI-BLAST cannot. Phyre2 has been designed to ensure a user-friendly interface for users inexpert in protein structure prediction methods. Its development is funded by the Biotechnology and Biological Sciences Research Council.

Macromolecular structure validation is the process of evaluating reliability for 3-dimensional atomic models of large biological molecules such as proteins and nucleic acids. These models, which provide 3D coordinates for each atom in the molecule, come from structural biology experiments such as x-ray crystallography or nuclear magnetic resonance (NMR). The validation has three aspects: 1) checking on the validity of the thousands to millions of measurements in the experiment; 2) checking how consistent the atomic model is with those experimental data; and 3) checking consistency of the model with known physical and chemical properties.
Michael Joseph Ezra Sternberg is a professor at Imperial College London, where he is director of the Centre for Integrative Systems Biology and Bioinformatics and Head of the Structural bioinformatics Group.

Transmembrane protein 171 (TMEM171) is a protein that in humans is encoded by the TMEM171 gene.