
Structural bioinformatics is the branch of bioinformatics that is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA. It deals with generalizations about macromolecular 3D structures such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, binding interactions, and structure/function relationships, working both from experimentally solved structures and from computational models. The term structural has the same meaning as in structural biology, and structural bioinformatics can be seen as a part of computational structural biology. The main objective of structural bioinformatics is the creation of new methods of analysing and manipulating biological macromolecular data in order to solve problems in biology and generate new knowledge.
BioJava is an open-source software project dedicated to provide Java tools to process biological data. BioJava is a set of library functions written in the programming language Java for manipulating sequences, protein structures, file parsers, Common Object Request Broker Architecture (CORBA) interoperability, Distributed Annotation System (DAS), access to AceDB, dynamic programming, and simple statistical routines. BioJava supports a huge range of data, starting from DNA and protein sequences to the level of 3D protein structures. The BioJava libraries are useful for automating many daily and mundane bioinformatics tasks such as to parsing a Protein Data Bank (PDB) file, interacting with Jmol and many more. This application programming interface (API) provides various file parsers, data models and algorithms to facilitate working with the standard data formats and enables rapid application development and analysis.
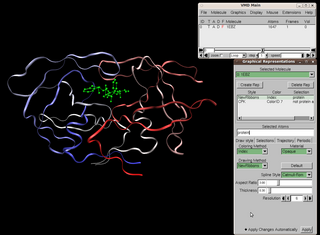
Visual Molecular Dynamics (VMD) is a molecular modelling and visualization computer program. VMD is developed mainly as a tool to view and analyze the results of molecular dynamics simulations. It also includes tools for working with volumetric data, sequence data, and arbitrary graphics objects. Molecular scenes can be exported to external rendering tools such as POV-Ray, RenderMan, Tachyon, Virtual Reality Modeling Language (VRML), and many others. Users can run their own Tcl and Python scripts within VMD as it includes embedded Tcl and Python interpreters. VMD runs on Unix, Apple Mac macOS, and Microsoft Windows. VMD is available to non-commercial users under a distribution-specific license which permits both use of the program and modification of its source code, at no charge.

Molecular modelling encompasses all methods, theoretical and computational, used to model or mimic the behaviour of molecules. The methods are used in the fields of computational chemistry, drug design, computational biology and materials science to study molecular systems ranging from small chemical systems to large biological molecules and material assemblies. The simplest calculations can be performed by hand, but inevitably computers are required to perform molecular modelling of any reasonably sized system. The common feature of molecular modelling methods is the atomistic level description of the molecular systems. This may include treating atoms as the smallest individual unit, or explicitly modelling protons and neutrons with its quarks, anti-quarks and gluons and electrons with its photons.
Q-Chem is a general-purpose electronic structure package featuring a variety of established and new methods implemented using innovative algorithms that enable fast calculations of large systems on various computer architectures, from laptops and regular lab workstations to midsize clusters and HPCC, using density functional and wave-function based approaches. It offers an integrated graphical interface and input generator; a large selection of functionals and correlation methods, including methods for electronically excited states and open-shell systems; solvation models; and wave-function analysis tools. In addition to serving the computational chemistry community, Q-Chem also provides a versatile code development platform.

PyMOL is an open source but proprietary molecular visualization system created by Warren Lyford DeLano. It was commercialized initially by DeLano Scientific LLC, which was a private software company dedicated to creating useful tools that become universally accessible to scientific and educational communities. It is currently commercialized by Schrödinger, Inc. As the original software license was a permissive licence, they were able to remove it; new versions are no longer released under the Python license, but under a custom license, and some of the source code is no longer released. PyMOL can produce high-quality 3D images of small molecules and biological macromolecules, such as proteins. According to the original author, by 2009, almost a quarter of all published images of 3D protein structures in the scientific literature were made using PyMOL.

Ghemical is a computational chemistry software package written in C++ and released under the GNU General Public License. The program has graphical user interface based on GTK+2 and supports quantum mechanical and molecular mechanic models, with geometry optimization, molecular dynamics, and a large set of visualization tools. Ghemical relies on external code to provide the quantum-mechanical calculations — MOPAC provides the semi-empirical MNDO, MINDO, AM1, and PM3 methods, and MPQC methods based on Hartree–Fock calculations.
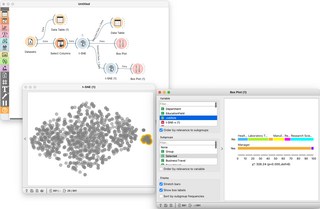
Orange is an open-source data visualization, machine learning and data mining toolkit. It features a visual programming front-end for explorative qualitative data analysis and interactive data visualization.

The Visualization Toolkit (VTK) is an open-source software system for 3D computer graphics, image processing and scientific visualization.
Graph Modeling Language (GML) is a hierarchical ASCII-based file format for describing graphs. It has been also named Graph Meta Language.
General Atomic and Molecular Electronic Structure System (GAMESS (US)) is computer software for computational chemistry. The original code started on October 1, 1977 as a National Resources for Computations in Chemistry project. In 1981, the code base split into GAMESS (US) and GAMESS (UK) variants, which now differ significantly. GAMESS (US) is maintained by the members of the Gordon Research Group at Iowa State University. GAMESS (US) source code is available as source-available freeware, but is not open-source software, due to license restrictions.
The Molecular Modelling Toolkit (MMTK) is an open-source software package written in Python, which performs common tasks in molecular modelling.
The Molecular Modeling Toolkit is a library that implements common molecular simulation techniques, with an emphasis on biomolecular simulations. It uses modern software engineering techniques in order to overcome limitations associated with the large monolithic simulation programs that are commonly used for biomolecules. Its principal advantages are (1) easy extension and combination with other libraries due to modular library design, (2) a single high-level general-purpose programming language (Python) is used for library implementation as well as for application scripts, (3) use of documented and machine-independent formats for all data files, and (4) interfaces to other simulation and visualization programs.
OpenMS is an open-source project for data analysis and processing in mass spectrometry and is released under the 3-clause BSD licence. It supports most common operating systems including Microsoft Windows, MacOS and Linux.

Ascalaph Designer is a computer program for general purpose molecular modelling for molecular design and simulations. It provides a graphical environment for the common programs of quantum and classical molecular modelling ORCA, NWChem, Firefly, CP2K and MDynaMix . The molecular mechanics calculations cover model building, energy optimizations and molecular dynamics. Firefly covers a wide range of quantum chemistry methods. Ascalaph Designer is free and open-source software, released under the GNU General Public License, version 2 (GPLv2).
Discovery Studio is a suite of software for simulating small molecule and macromolecule systems. It is developed and distributed by Dassault Systemes BIOVIA.
Smoldyn is an open-source software application for cell-scale biochemical simulations. It uses particle-based simulation, meaning that it simulates each molecule of interest individually, in order to capture natural stochasticity and yield nanometer-scale spatial resolution. Simulated molecules diffuse, react, are confined by surfaces, and bind to membranes in similar manners as in real biochemical systems.
