A chemical database is a database specifically designed to store chemical information. This information is about chemical and crystal structures, spectra, reactions and syntheses, and thermophysical data.
Cheminformatics refers to the use of physical chemistry theory with computer and information science techniques—so called "in silico" techniques—in application to a range of descriptive and prescriptive problems in the field of chemistry, including in its applications to biology and related molecular fields. Such in silico techniques are used, for example, by pharmaceutical companies and in academic settings to aid and inform the process of drug discovery, for instance in the design of well-defined combinatorial libraries of synthetic compounds, or to assist in structure-based drug design. The methods can also be used in chemical and allied industries, and such fields as environmental science and pharmacology, where chemical processes are involved or studied.
Chemical Markup Language is an approach to managing molecular information using tools such as XML and Java. It was the first domain specific implementation based strictly on XML, first based on a DTD and later on an XML Schema, the most robust and widely used system for precise information management in many areas. It has been developed over more than a decade by Murray-Rust, Rzepa and others and has been tested in many areas and on a variety of machines.
Quantitative structure–activity relationship models are regression or classification models used in the chemical and biological sciences and engineering. Like other regression models, QSAR regression models relate a set of "predictor" variables (X) to the potency of the response variable (Y), while classification QSAR models relate the predictor variables to a categorical value of the response variable.
A molecule editor is a computer program for creating and modifying representations of chemical structures.
The International Chemical Identifier is a textual identifier for chemical substances, designed to provide a standard way to encode molecular information and to facilitate the search for such information in databases and on the web. Initially developed by the International Union of Pure and Applied Chemistry (IUPAC) and National Institute of Standards and Technology (NIST) from 2000 to 2005, the format and algorithms are non-proprietary. Since May 2009, it has been developed by the InChI Trust, a nonprofit charity from the United Kingdom which works to implement and promote the use of InChI.

Open Babel is computer software, a chemical expert system mainly used to interconvert chemical file formats.

Jmol is computer software for molecular modelling chemical structures in 3-dimensions. Jmol returns a 3D representation of a molecule that may be used as a teaching tool, or for research e.g., in chemistry and biochemistry. It is written in the programming language Java, so it can run on the operating systems Windows, macOS, Linux, and Unix, if Java is installed. It is free and open-source software released under a GNU Lesser General Public License (LGPL) version 2.0. A standalone application and a software development kit (SDK) exist that can be integrated into other Java applications, such as Bioclipse and Taverna.

JOELib is computer software, a chemical expert system used mainly to interconvert chemical file formats. Because of its strong relationship to informatics, this program belongs more to the category cheminformatics than to molecular modelling. It is available for Windows, Unix and other operating systems supporting the programming language Java. It is free and open-source software distributed under the GNU General Public License (GPL) 2.0.

The Chemistry Development Kit (CDK) is computer software, a library in the programming language Java, for chemoinformatics and bioinformatics. It is available for Windows, Linux, Unix, and macOS. It is free and open-source software distributed under the GNU Lesser General Public License (LGPL) 2.0.

Peter Murray-Rust is a chemist currently working at the University of Cambridge. As well as his work in chemistry, Murray-Rust is also known for his support of open access and open data.
BALL is a C++ class framework and set of algorithms and data structures for molecular modelling and computational structural bioinformatics, a Python interface to this library, and a graphical user interface to BALL, the molecule viewer BALLView.
ChemSpider is a freely accessible online database of chemicals owned by the Royal Society of Chemistry. It contains information on more than 100 million molecules from over 270 data sources, each of them receiving a unique identifier called ChemSpider Identifier.
Chemaxon is a cheminformatics and bioinformatics software development company, headquartered in Budapest with 250 employees. The company also has offices in Cambridge, San Diego, Basel and in Prague. and it has distributors in China, India, Japan, South Korea, Singapore, and Australia.

Chemical similarity refers to the similarity of chemical elements, molecules or chemical compounds with respect to either structural or functional qualities, i.e. the effect that the chemical compound has on reaction partners in inorganic or biological settings. Biological effects and thus also similarity of effects are usually quantified using the biological activity of a compound. In general terms, function can be related to the chemical activity of compounds.
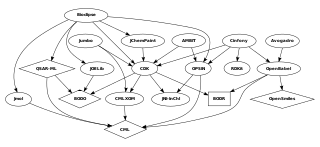
Blue Obelisk is an informal group of chemists who promote open data, open source, and open standards; it was initiated by Peter Murray-Rust and others in 2005. Multiple open source cheminformatics projects associate themselves with the Blue Obelisk, among which, in alphabetical order, Avogadro, Bioclipse, cclib, Chemistry Development Kit, GaussSum, JChemPaint, JOELib, Kalzium, Openbabel, OpenSMILES, and UsefulChem.

Avogadro is a molecule editor and visualizer designed for cross-platform use in computational chemistry, molecular modeling, bioinformatics, materials science, and related areas. It is extensible via a plugin architecture.

The Journal of Cheminformatics is a peer-reviewed open access scientific journal that covers cheminformatics and molecular modelling. It was established in 2009 with David Wild and Christoph Steinbeck as founding editors-in-chief, and was originally published by Chemistry Central. At the end of 2015, the Chemistry Central brand was retired and its titles, including Journal of Cheminformatics, were merged with the SpringerOpen portfolio of open access journals.
A chemical graph generator is a software package to generate computer representations of chemical structures adhering to certain boundary conditions. The development of such software packages is a research topic of cheminformatics. Chemical graph generators are used in areas such as virtual library generation in drug design, in molecular design with specified properties, called inverse QSAR/QSPR, as well as in organic synthesis design, retrosynthesis or in systems for computer-assisted structure elucidation (CASE). CASE systems again have regained interest for the structure elucidation of unknowns in computational metabolomics, a current area of computational biology.
