
Fanconi anemia (FA) is a rare, AR, genetic disease resulting in impaired response to DNA damage in the FA/BRCA pathway. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia (AML), MDS, and liver tumors. 90% develop aplastic anemia by age 40. About 60–75% have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% have some form of endocrine problem, with varying degrees of severity. 60% of FA is FANC-A, 16q24.3, which has later onset bone marrow failure.
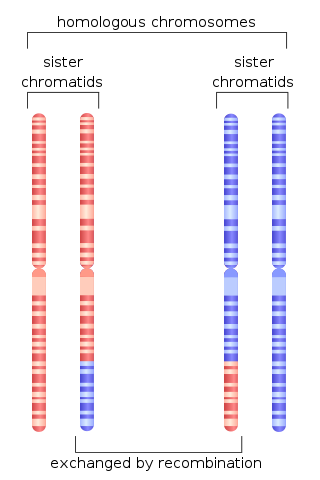
Homologous recombination is a type of genetic recombination in which genetic information is exchanged between two similar or identical molecules of double-stranded or single-stranded nucleic acids.
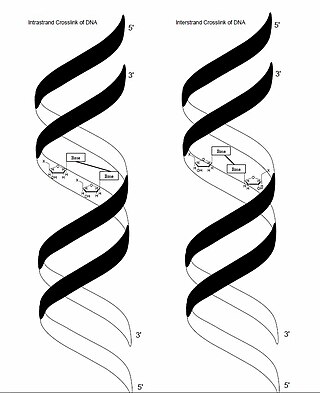
In genetics, crosslinking of DNA occurs when various exogenous or endogenous agents react with two nucleotides of DNA, forming a covalent linkage between them. This crosslink can occur within the same strand (intrastrand) or between opposite strands of double-stranded DNA (interstrand). These adducts interfere with cellular metabolism, such as DNA replication and transcription, triggering cell death. These crosslinks can, however, be repaired through excision or recombination pathways.

DNA excision repair protein ERCC-1 is a protein that in humans is encoded by the ERCC1 gene. Together with ERCC4, ERCC1 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.

Fanconi anemia group C protein is a protein that in humans is encoded by the FANCC gene. This protein delays the onset of apoptosis and promotes homologous recombination repair of damaged DNA. Mutations in this gene result in Fanconi anemia, a human rare disorder characterized by cancer susceptibility and cellular sensitivity to DNA crosslinks and other damages.

Fanconi anemia group D2 protein is a protein that in humans is encoded by the FANCD2 gene. The Fanconi anemia complementation group (FANC) currently includes FANCA, FANCB, FANCC, FANCD1, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN and FANCO.
Fanconi anemia group G protein is a protein that in humans is encoded by the FANCG gene.
ERCC4 is a protein designated as DNA repair endonuclease XPF that in humans is encoded by the ERCC4 gene. Together with ERCC1, ERCC4 forms the ERCC1-XPF enzyme complex that participates in DNA repair and DNA recombination.

E3 ubiquitin-protein ligase FANCL is an enzyme that in humans is encoded by the FANCL gene.

Fanconi anemia group B protein is a protein that in humans is encoded by the FANCB gene.

Fanconi anemia, complementation group I (FANCI) also known as KIAA1794, is a protein which in humans is encoded by the FANCI gene. Mutations in the FANCI gene are known to cause Fanconi anemia.

Partner and localizer of BRCA2, also known as PALB2 or FANCN, is a protein which in humans is encoded by the PALB2 gene.

RecQ-mediated genome instability protein 1 is a protein that in humans is encoded by the RMI1 gene.

Fanconi anemia, complementation group M, also known as FANCM is a human gene. It is an emerging target in cancer therapy, in particular cancers with specific genetic deficiencies.
Sgs1, also known as slow growth suppressor 1, is a DNA helicase protein found in Saccharomyces cerevisiae. It is a homolog of the bacterial RecQ helicase. Like the other members of the RecQ helicase family, Sgs1 is important for DNA repair. In particular, Sgs1 collaborates with other proteins to repair double-strand breaks during homologous recombination in eukaryotes.
FANC proteins are a network of at least 15 proteins that are associated with a cell process known as the Fanconi anemia.
FANCD2/FANCI-associated nuclease 1 (KIAA1018) is an enzyme that in humans is encoded by the FAN1 gene. It is a structure dependent endonuclease. It is thought to play an important role in the Fanconi Anemia (FA) pathway.

SLX4 interacting protein is a protein that in humans is encoded by the SLX4IP gene.
Agata Smogorzewska is a Polish-born scientist. She is an associate professor at Rockefeller University, heading the Laboratory of Genome Maintenance. Her work primarily focuses on DNA interstrand crosslink repair and the diseases resulting from deficiencies in this repair pathway, including Fanconi anemia and karyomegalic interstitial nephritis.

A double-strand break repair model refers to the various models of pathways that cells undertake to repair double strand-breaks (DSB). DSB repair is an important cellular process, as the accumulation of unrepaired DSB could lead to chromosomal rearrangements, tumorigenesis or even cell death. In human cells, there are two main DSB repair mechanisms: Homologous recombination (HR) and non-homologous end joining (NHEJ). HR relies on undamaged template DNA as reference to repair the DSB, resulting in the restoration of the original sequence. NHEJ modifies and ligates the damaged ends regardless of homology. In terms of DSB repair pathway choice, most mammalian cells appear to favor NHEJ rather than HR. This is because the employment of HR may lead to gene deletion or amplification in cells which contains repetitive sequences. In terms of repair models in the cell cycle, HR is only possible during the S and G2 phases, while NHEJ can occur throughout whole process. These repair pathways are all regulated by the overarching DNA damage response mechanism. Besides HR and NHEJ, there are also other repair models which exists in cells. Some are categorized under HR, such as synthesis-dependent strain annealing, break-induced replication, and single-strand annealing; while others are an entirely alternate repair model, namely, the pathway microhomology-mediated end joining (MMEJ).



