
Muscular dystrophies (MD) are a genetically and clinically heterogeneous group of rare neuromuscular diseases that cause progressive weakness and breakdown of skeletal muscles over time. The disorders differ as to which muscles are primarily affected, the degree of weakness, how fast they worsen, and when symptoms begin. Some types are also associated with problems in other organs.
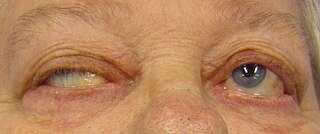
Myasthenia gravis (MG) is a long-term neuromuscular junction disease that leads to varying degrees of skeletal muscle weakness. The most commonly affected muscles are those of the eyes, face, and swallowing. It can result in double vision, drooping eyelids, and difficulties in talking and walking. Onset can be sudden. Those affected often have a large thymus or develop a thymoma.
The muscular system is an organ system consisting of skeletal, smooth, and cardiac muscle. It permits movement of the body, maintains posture, and circulates blood throughout the body. The muscular systems in vertebrates are controlled through the nervous system although some muscles can be completely autonomous. Together with the skeletal system in the human, it forms the musculoskeletal system, which is responsible for the movement of the body.

Limb–girdle muscular dystrophy (LGMD) is a genetically heterogeneous group of rare muscular dystrophies that share a set of clinical characteristics. It is characterised by progressive muscle wasting which affects predominantly hip and shoulder muscles. LGMD usually has an autosomal pattern of inheritance. It currently has no known cure or treatment.

Myostatin is a protein that in humans is encoded by the MSTN gene. Myostatin is a myokine that is produced and released by myocytes and acts on muscle cells to inhibit muscle growth. Myostatin is a secreted growth differentiation factor that is a member of the TGF beta protein family.
Muscular Dystrophy Association (MDA) is an American nonprofit organization dedicated to supporting people living with muscular dystrophy, ALS, and related neuromuscular diseases. Founded in 1950 by Paul Cohen, who lived with muscular dystrophy, MDA accelerates research, advances care, and works to empower families to live longer and more independent lives. Renowned for The MDA Labor Day Telethon, the annual telecast aired live every Labor Day weekend, with comedian and filmmaker Jerry Lewis as its host and national chairman. Don Rickles, Frank Sinatra, Sammy Davis Jr., Milton Berle, Wayne Newton, Norm Crosby, Don Francisco, Tony Orlando, Johnny Carson, Aretha Franklin, Maureen McGovern, Diana Ross, Angela Lansbury and others have supported MDA over the years. The organization's headquarters is in Chicago, Illinois.
Dystrophin is a rod-shaped cytoplasmic protein, and a vital part of a protein complex that connects the cytoskeleton of a muscle fiber to the surrounding extracellular matrix through the cell membrane. This complex is variously known as the costamere or the dystrophin-associated protein complex (DAPC). Many muscle proteins, such as α-dystrobrevin, syncoilin, synemin, sarcoglycan, dystroglycan, and sarcospan, colocalize with dystrophin at the costamere. It has a molecular weight of 427 kDa

Duchenne muscular dystrophy (DMD) is a severe type of muscular dystrophy predominantly affecting boys. The onset of muscle weakness typically begins around age four, with rapid progression. Initially, muscle loss occurs in the thighs and pelvis, extending to the arms, which can lead to difficulties in standing up. By the age of 12, most individuals with Duchenne muscular dystrophy are unable to walk. Affected muscles may appear larger due to an increase in fat content, and scoliosis is common. Some individuals may experience intellectual disability, and females carrying a single copy of the mutated gene may show mild symptoms.

Becker muscular dystrophy (BMD) is an X-linked recessive inherited disorder characterized by slowly progressing muscle weakness of the legs and pelvis. It is a type of dystrophinopathy. The cause is mutations and deletions in any of the 79 exons encoding the large dystrophin protein, essential for maintaining the muscle fiber's cell membrane integrity. Becker muscular dystrophy is related to Duchenne muscular dystrophy in that both result from a mutation in the dystrophin gene, however the hallmark of Becker is milder in-frame deletions. and hence has a milder course, with patients maintaining ambulation till 50–60 years if detected early.
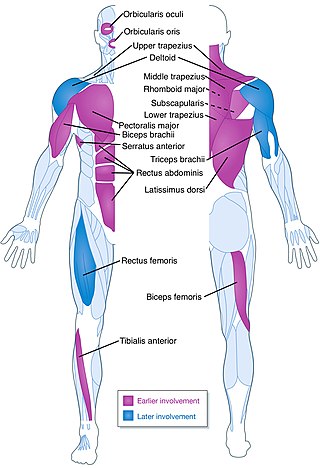
Facioscapulohumeral muscular dystrophy (FSHD) is a type of muscular dystrophy, a group of heritable diseases that cause degeneration of muscle and progressive weakness. Per the name, FSHD tends to sequentially weaken the muscles of the face, those that position the scapula, and those overlying the humerus bone of the upper arm. These areas can be spared, and muscles of other areas usually are affected, especially those of the chest, abdomen, spine, and shin. Almost any skeletal muscle can be affected in advanced disease. Abnormally positioned, termed 'winged', scapulas are common, as is the inability to lift the foot, known as foot drop. The two sides of the body are often affected unequally. Weakness typically manifests at ages 15–30 years. FSHD can also cause hearing loss and blood vessel abnormalities at the back of the eye.

Omigapil is a drug that was developed by Novartis and tested in clinical trials for its ability to help treat Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS). The development for PD and ALS have been terminated due to lack of benefit, but Santhera Pharmaceuticals bought the compound for development for the treatment of congenital muscular dystrophy (CMD).

Spinal muscular atrophy (SMA) is a rare neuromuscular disorder that results in the loss of motor neurons and progressive muscle wasting. It is usually diagnosed in infancy or early childhood and if left untreated it is the most common genetic cause of infant death. It may also appear later in life and then have a milder course of the disease. The common feature is progressive weakness of voluntary muscles, with arm, leg and respiratory muscles being affected first. Associated problems may include poor head control, difficulties swallowing, scoliosis, and joint contractures.
Follistatin, also known as activin-bindings protein, is a protein that in humans is encoded by the FST gene. Follistatin is an autocrine glycoprotein that is expressed in nearly all tissues of higher animals.
The activin type 2 receptors belong to a larger TGF-beta receptor family and modulate signals for transforming growth factor beta ligands. These receptors are involved in a host of physiological processes including, growth, cell differentiation, homeostasis, osteogenesis, apoptosis and many other functions. There are two activin type two receptors: ACVR2A and ACVR2B.

Pikachurin, also known as AGRINL (AGRINL) and EGF-like, fibronectin type-III and laminin G-like domain-containing protein (EGFLAM), is a protein that in humans is encoded by the EGFLAM gene.
Sunil Pradhan is an Indian neurologist, medical researcher and writer, known for the invention of two electrophysiological techniques. He has also described five medical signs, of which one related to Duchenne muscular dystrophy is known as Pradhan Sign, and the others associated with facioscapulohumeral muscular dystrophy (FSHD) and similar neuro diseases. The Government of India awarded him the Padma Shri, the fourth highest civilian award, in 2014 for his contributions to the field of neuroscience.

A decoy receptor is a receptor that is able to recognize and bind specific growth factors or cytokines efficiently, but is not structurally able to signal or activate the intended receptor complex. It acts as an inhibitor, binding a ligand and keeping it from binding to its regular receptor. Decoy receptors participate in a common methods of signal inhibition and are also abundant in malignant tissues, making up a significant topic in cancer research.

Casimersen, sold under the brand name Amondys 45, is an antisense oligonucleotide medication used for the treatment of Duchenne muscular dystrophy (DMD) in people who have a confirmed mutation of the dystrophin gene that is amenable to exon 45 skipping. It is an antisense oligonucleotide of phosphorodiamidate morpholino oligomer (PMO). Duchenne muscular dystrophy is a rare disease that primarily affects boys. It is caused by low levels of a muscle protein called dystrophin. The lack of dystrophin causes progressive muscle weakness and premature death.
Myostatin inhibitors are a class of drugs that work by blocking the effects of myostatin, which inhibits muscle growth. In animal models and limited human studies, myostatin inhibitors have increased muscle size. They are being developed to treat obesity, sarcopenia, muscular dystrophy, and other illnesses.
Apitegromab (SRK-015) is a fully human monoclonal antibody developed to treat spinal muscular atrophy. It works by binding to and inhibiting promyostatin, a precursor to myostatin, which limits the size of skeletal muscle tissue, as well as inactive myostatin. It does not bind to active myostatin, activin A, active BMP9/10 or TGFβ1 that all operate on the activin type 2 receptors.