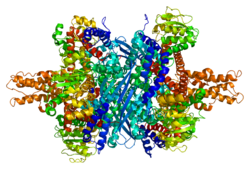
The color scheme is as follows: Glu-BD, NAD(P)-BD, antena, the pivot helix
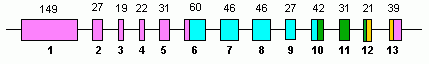
Human GLUD1 contains 13 exons and is located on the 10th chromosome.
There is evidence that GLUD1 has been retro-posed to the X chromosome, where it gave rise to the intronless GLUD2 through random mutations and natural selection. GLUD2 have adapted to the particular needs of the nervous system where it is specifically expressed.[5]
Protein
The domain structure of GLUD1 Each domain is colored differently - Glu-BD, NAD(P)-BD, antenna, the pivot helix. The allosteric regulators are shown as sphere models. This particular structure of GLUD1 is a combination of two X-ray structures - one with a bound GTP (1HWZ) and the second one with a bound ADP (1NQT,8AR8). Although not real, this structure shows the relative position of the allosteric effectors when bound to GLUD1. NADPH and Glu are shown as well.
GLUD1 is a hexamer. The monomer unit has:
N-terminal Glu-BD(Binding domain) that is composed mostly of β-strands.
NAD-BD - can bind either NAD+ or NADP+.
48-residue antenna-like projection that extends from the top of each NAD-BD. The antenna consists of an ascending helix and a descending random coil strand that contains a small α-helix toward the C-terminal end of the strand.
NAD-BD sits on the top of Glu-BD. NAD-BD and Glu-BD form the catalytic cleft. During substrate binding, the NAD-BD moves significantly. This movement has two components, rotating along the long axis of a helix at the back of the NAD-BD, called "the pivot helix", and twisting about the antenna in a clockwise fashion. A comparison of the open and closed conformations of GLUD1 reveals changes in the small helix of the descending strand of the antenna, which seems to recoil as the catalytic cleft opens.[6] Closure of one subunit is associated with distortion of the small helix of the descending strand that is pushed into the antenna of the adjacent subunit. R496 is located on this small helix (see Mutations).
The core structure of the hexamer is a stacked dimer of trimers. Glu-BDs of the monomers are mainly responsible in the buildup of the core. The relative position of the monomers is such that the rotation about the pivot helix in each monomer is not restricted. The antennae from three subunits within the trimers wrap around each other and undergo conformational changes as the catalytic cleft opens and closes. The antenna serves as an intersubunit communication conduit during negative cooperativity and allosteric regulation.
Alignment of GLUD1 from various sources, shows that the antenna probably evolved in the protista prior to the formation of purine regulatory sites. This suggests that there is some selective advantage of the antenna itself and that animals evolved new functions for GLUD1 through the addition of allosteric regulation.[7]
GLUD1 can form long fibers by end to end association of the hexamers. The polymerization is unrelated to the catalytic activity, but probably has an important role such as formation of multienzyme complexes.
GLUD1 has two co-enzyme binding sites: one in the NAD-BD that is able to bind ether NAD+ or NADP+ and is directly involved in the catalytic process, and a second one, that has regulatory function, lying directly under the pivot helix, that can bind ADP, NAD+, or NADH, but does not bind NADPH well.[8]
Function
GLUD1 catalyses the oxidative deamination of Glu to 2-oxoglutarate and free NH4+ using either NAD+ or NADP+ as a co-factor. The reaction occurs with the transfer of a hydride ion from Glu's Cα to NAD(P)+, thereby forming 2-iminoglutarate, which is hydrolyzed to 2-oxoglutarate and NH4+. The reaction's equilibrium under standard circumstances greatly favors Glu formation over NH4+ (Go' ~ 30 kJ.mol-1) formation. For this reason, it was thought that the enzyme played an important role in ammonia detoxification, because since high [NH4+] are toxic, this equilibrium position would be physiologically important; it would help to maintain low [NH4+]. However, in individuals with a certain form of hyperammonemia resulting from a form of hyperinsulinism, the enzyme's activity is increased due to decreased GTP sensitivity, a negative regulator. These individual's blood ammonia levels are raised significantly, which would not be expected if the enzyme did indeed operate at equilibrium.
Interactions
Binding partners
ADP
ADP binds behind the NAD-BD, just beneath the pivot helix - in the special ADP allosteric site. The adenosine moiety binds down into a hydrophobic pocket, with the ribose phosphate groups pointing outside towards the GTP allosteric site.[9]
NADH can also bind to the ADP site, when at high concentrations, usually resulting in the enzyme inhibition.[10]
GTP
GTP binding is antagonized by Pi and ADP but is synergistic with NADH bound in the noncatalytic allosteric site. The majority of the contacts between GTP and the enzyme are via the triphosphate moiety. The GTP-binding site is considered to be the "sensor" that turns the enzyme off when the cell is at a high energy state. GTP binds at the junction between the NAD-BD and the antenna.[8][11]
Whereas most of the GLUD1-GTP interactions are via β- and γ-phosphate interactions, there are specific interactions with E346 and K343 that favour guanosine over adenosine.
In the open conformation, the GTP binding site is distorted such that it can no longer bind GTP.[6]
Regulation
When GLUD1 is highly saturated with the active site ligands (substrates), an inhibitory abortive complex forms in the active site: NAD(P)H.Glu in the oxidative deamination reaction at high pH, and NAD(P)+.2-oxoglutarate in the reductive amination reaction at low pH. GLUD1 assumes its basal state configuration in the absence of allosteric effectors, regardless of whether the allosteric sites are functional. The allosteric regulators of GLUD1 - ADP, GTP, Leu, NAD+ and NADH - exert their effects by changing the energy required to open and close the catalytic cleft during enzymic turnover, in other words by destabilizing or stabilizing, respectively, the abortive complexes. Activators are not necessary for the catalytic function of GLUD1, as it is active in the absence of these compounds (basal state). It has been suggested that GLUD1 assumes in its basal state a configuration (open catalytic cleft) that permits catalytic activity regardless of whether the allosteric sites are functional. GLUD regulation is of particular biological importance as exemplified by observations showing that regulatory mutations of GLUD1 are associated with clinical manifestations in children.
ADP
ADP being one of the two major activators (NAD+ being the other one), acts by destabilizing the abortive complexes, and abrogating the negative cooperativity. In the absence of substrates, and with bound ADP, the catalytic cleft is in the open conformation, and the GLUD1 hexamers form long polymers in the crystal cell with more interactions than found in the abortive complex crystals (8AR8[9]). This is consistent with the fact that ADP promotes aggregation in solution. When the catalytic cleft opens, R516 is rotated down on to the phosphates of ADP.[8] The opening of the catalytic cleft is roughly correlated with distance between R516 and phosphates of ADP. In this way, ADP activates GLUD1 by facilitating the opening of the catalytic cleft which decreases product affinity and facilitates product release.[6][12] thus allowing GLUD1 to reconcile the non-catalytic abortive complexes.[11]
Inhibition by high [ADP] has been suggested previously to be due to competition between ADP and the adenosine moiety of the coenzyme at the active site1. At least it is known that the effect is relatively unaffected by either H507Y or R516A.
ATP
ATP has complex concentration dependent effects on GLUD1 activity:
Low [ATP] - inhibition, mediated through the GTP-binding site, since it is eliminated by H507Y. The affinity of ATP for the GTP site appears to be 1000-fold lower than for GTP, since the β- and γ-phosphate interactions are the major determinant of binding at the GTP site.
Intermediate [ATP] - activation, mediated through the ADP effector site, since it is almost completely eliminated by R516A. At this site the nucleotide group is the major determinant of binding.
High [ATP] - inhibition, mediated by weak binding at a third site, which is relatively specific for the adenine nucleotides. This effect is relatively unaffected by either H507Y or R516A. As suggested for ADP it could be due to a competition between ATP and the adenosine moiety of the coenzyme at the active site.[13]
GTP
GTP inhibits enzyme turnover over a wide range of conditions by increasing the affinity of GLUD1 for the reaction product, making product release rate limiting under all conditions in the presence of GTP. GTP acts by keeping the catalytic cleft in a closed conformation thus stabilizing the abortive complexes. GTP effects on GLUD1 are not localized solely to the subunit to which it is binding and that the antenna plays an important role in communicating this inhibition to other subunits.
Leu
Leu activates GLUD1 independently of the ADP, possessing a special allosteric site in the subunits interface area 8AR7.[9] The enhanced responses of HI/HA patients (see HI/HA syndrom) to Leu stimulation of INS release3, which result from their impaired sensitivity to GTP inhibition, emphasize the physiological importance of inhibitory control of GLUD1.[13]
NAD+
NAD(P)(H) can bind to a second site on each subunit. This site binds NAD(H) ~ 10-fold better than NADP(H) with the reduced forms better than the oxidized forms.[10] Although it has been suggested that binding of the reduced coenzyme at this site inhibits the reaction, while oxidized coenzyme binding causes activation, the effect is still unclear.
NADH
NADH, is another major allosteric inhibitor of GLUD1.
Phosphate
Phosphate and other bivalent anions stabilize GLUD1. Recent structural studies have shown that phosphate molecules bind to the GTP site.[8]
Clinical significance
Familial hyperinsulinism, linked to mutations in GLUD1, is characterized by hypoglycemia that ranges from severe neonatal-onset, difficult-to-manage disease to childhood-onset disease with mild symptoms and difficult-to-diagnose hypoglycemia. Neonatal-onset disease manifests within hours to two days after birth. Childhood-onset disease manifests during the first months or years of life. In the newborn period, presenting symptoms may be nonspecific, including seizures, hypotonia, poor feeding, and apnea. In severe cases, serum glucose concentrations are typically extremely low and thus easily recognized, whereas in milder cases, variable and mild hypoglycemia may make the diagnosis more difficult. Even within the same family, disease manifestations can range from mild to severe. Individuals with autosomal recessive familial hyperinsulinism, caused by mutations in either ABCC8 or KCNJ11 (FHI-KATP), tend to be large for gestational age and usually present with severe refractory hypoglycemia in the first 48 hours of life; affected infants usually respond only partially to diet or medical management (i.e., diazoxide therapy) and thus may require pancreatic resection. Individuals with autosomal dominant FHI-KATP tend to be appropriate for gestational age at birth, to present at approximately age one year (range: 2 days - 30 years), and to respond to diet and diazoxide therapy. Exceptions to both of these generalities have been reported. FHI-GCK, caused by mutations in GCK, may be much milder than FHI-KATP; however, some persons have severe, diazoxide-unresponsive hypoglycemia. FHI-HADH, caused by mutations in HADH, tends to be relatively mild, although severe cases have been reported. Individuals with FHI-HNF4A, caused by mutations in HNF4A, are typically born large for gestational age and have mild features that respond to diazoxide treatment. FHI-UCP2, caused by mutations in UCP2, is a rare cause of diazoxide-responsive FH1. Hyperammonemia/hyperinsulinism (HA/HI) is associated with mild-to-moderate hyperammonemia and with relatively mild, late-onset hypoglycemia; most but not all affected individuals have mutations in GLUD1.[14]
Clinical characteristics
FHI is characterized by hypoglycemia that ranges from severe neonatal-onset, difficult-to-manage disease to childhood-onset disease with mild symptoms and difficult-to-diagnose hypoglycemia. Neonatal-onset disease manifests within hours to two days after birth. Childhood-onset disease manifests during the first months or years of life.[15] In the newborn period, presenting symptoms may be nonspecific, including seizures, hypotonia, poor feeding, and apnea. In severe cases, serum glucose concentrations are typically extremely low and thus easily recognized, whereas in milder cases, variable and mild hypoglycemia may make the diagnosis more difficult. Even within the same family, disease manifestations can range from mild to severe.[16]
Diagnosis/testing
Approximately 45% of affected individuals have mutations in either ABCC8, which encodes the protein SUR1, or KCNJ11, which encodes the protein Kir6.2. In the Ashkenazi Jewish population, two ABCC8 founder mutations are responsible for approximately 97% of FHI. Other ABCC8 founder mutations are present in the Finnish population (p.Val187Asp and p.Asp1506Lys). Mutations in GLUD1 and HNF4A each account for approximately 5% of individuals with FHI.[17][18] Activating mutations in GCK or inactivating mutations in HADH occur in fewer than 1% of individuals with FHI. Mutations in UCP2 have been reported in only two families to date. Approximately 40% of individuals with FHI do not have an identifiable mutation in any of the genes known to be associated with FHI.
Management
At initial diagnosis, hypoglycemia is corrected with intravenous glucose to normalize plasma glucose concentration and prevent brain damage.[19] Long-term medical management includes the use of diazoxide, somatostatin analogs, nifedipine, glucagon, recombinant IGF-I, glucocorticoids, human growth hormone, dietary intervention, or combinations of these therapies.[20] In individuals in whom aggressive medical management fails to maintain plasma glucose concentration within safe limits, or in whom such therapy cannot be safely maintained over time, pancreatic resection is considered.[21]
1 2 3 Smith TJ, Schmidt T, Fang J, Wu J, Siuzdak G, Stanley CA (May 2002). "The structure of apo human glutamate dehydrogenase details subunit communication and allostery". J. Mol. Biol. 318 (3): 765–77. doi:10.1016/S0022-2836(02)00161-4. PMID12054821.
↑ Banerjee S, Schmidt T, Fang J, Stanley CA, Smith TJ (April 2003). "Structural studies on ADP activation of mammalian glutamate dehydrogenase and the evolution of regulation". Biochemistry. 42 (12): 3446–56. doi:10.1021/bi0206917. PMID12653548.
1 2 3 4 Smith TJ, Peterson PE, Schmidt T, Fang J, Stanley CA (March 2001). "Structures of bovine glutamate dehydrogenase complexes elucidate the mechanism of purine regulation". J. Mol. Biol. 307 (2): 707–20. doi:10.1006/jmbi.2001.4499. PMID11254391.
↑ George A, Bell JE (December 1980). "Effects of adenosine 5'-diphosphate on bovine glutamate dehydrogenase: diethyl pyrocarbonate modification". Biochemistry. 19 (26): 6057–61. doi:10.1021/bi00567a017. PMID7470450.
↑ Won JG, Tseng HS, Yang AH, Tang KT, Jap TS, Lee CH, Lin HD, Burcus N, Pittenger G, Vinik A (Nov 2006). "Clinical features and morphological characterization of 10 patients with noninsulinoma pancreatogenous hypoglycaemia syndrome (NIPHS)". Clinical Endocrinology. 65 (5): 566–78. doi:10.1111/j.1365-2265.2006.02629.x. PMID17054456. S2CID19076202.
↑ Glaser B, Blech I, Krakinovsky Y, Ekstein J, Gillis D, Mazor-Aronovitch K, Landau H, Abeliovich D (Oct 2011). "ABCC8 mutation allele frequency in the Ashkenazi Jewish population and risk of focal hyperinsulinemic hypoglycemia". Genetics in Medicine. 13 (10): 891–4. doi:10.1097/GIM.0b013e31821fea33. PMID21716120. S2CID11352891.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.